
International Journal of Analytical and Bioanalytical Methods
(ISSN: 2633-8912)
Volume 1, Issue 1
Research Article
DOI: 10.35840/2633-8912/2405

Article Formats
Development and Validation of a High-Throughput Method for the Determination of Eight Non-Steroidal Anti-Inflammatory Drugs and Chloramphenicol in Milk, using Liquid Chromatography-Tandem Mass Spectroscopy
Table of Content
Figures
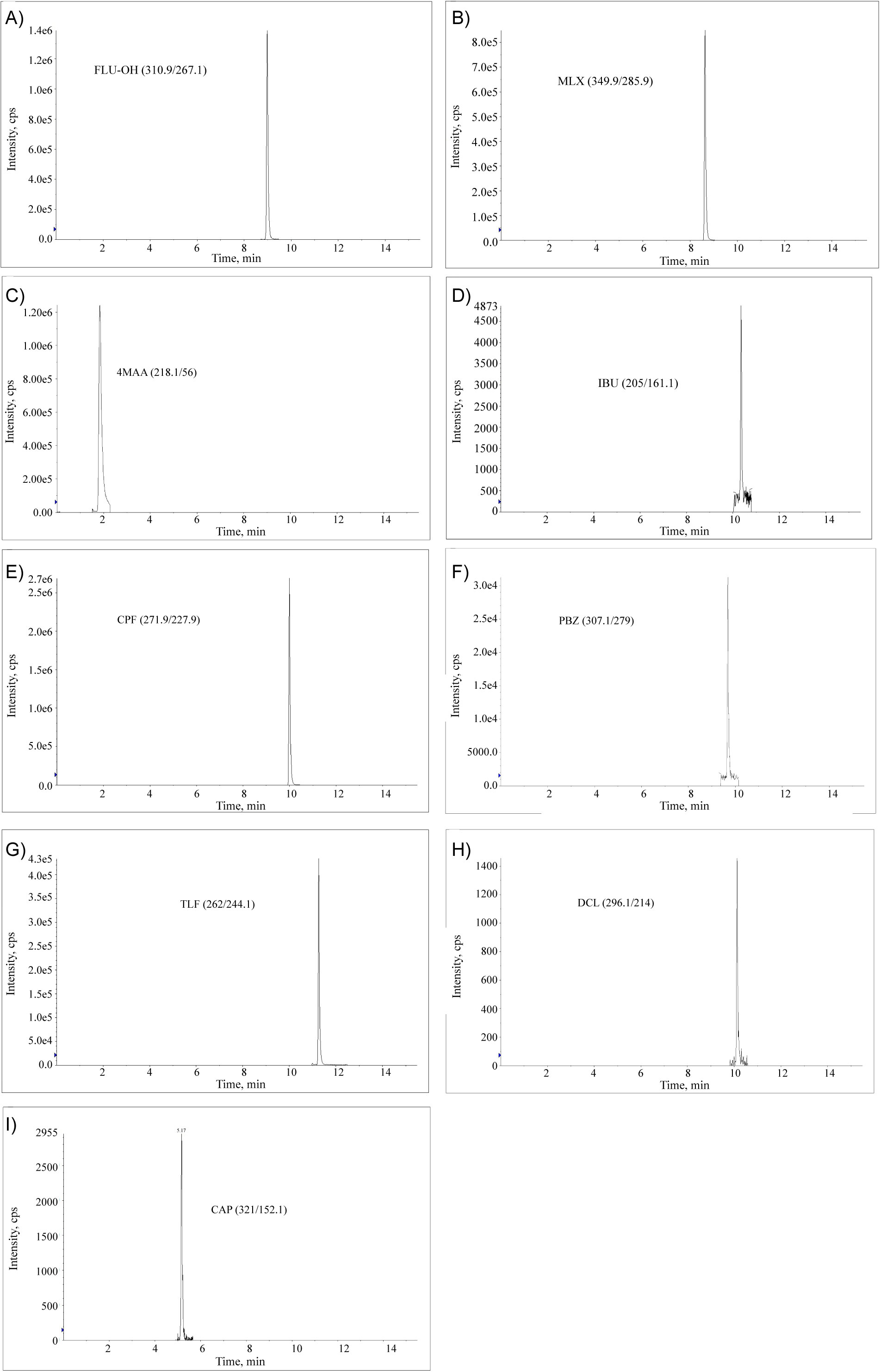
Figure 1: LC-MS/MS quantifier ion chromatograms...
LC-MS/MS quantifier ion chromatograms (sMRM mode) of a milk sample fortified at 0.5 MRL/RC of the studied analytes.

Figure 2: The implementation of the IBU...
The implementation of the IBU and DCL confirmation methods.
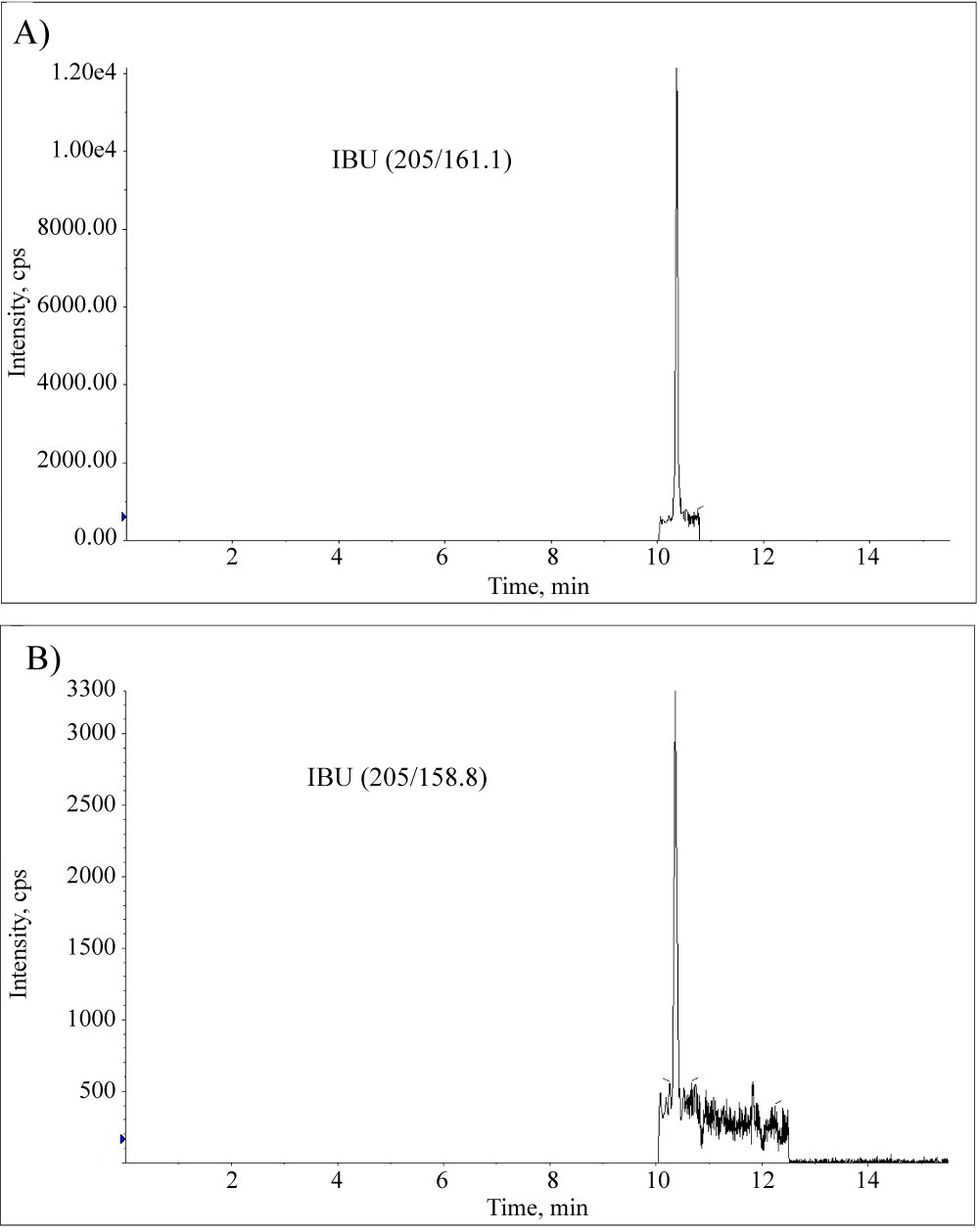
Figure 3: The quantifier (205/161.1) and qualifier...
The quantifier (205/161.1) and qualifier (205/158.8) ions chromatograms (sMRM scan) of IBU in an extracted milk sample spiked at 0.5 RC of 5 μg/kg.
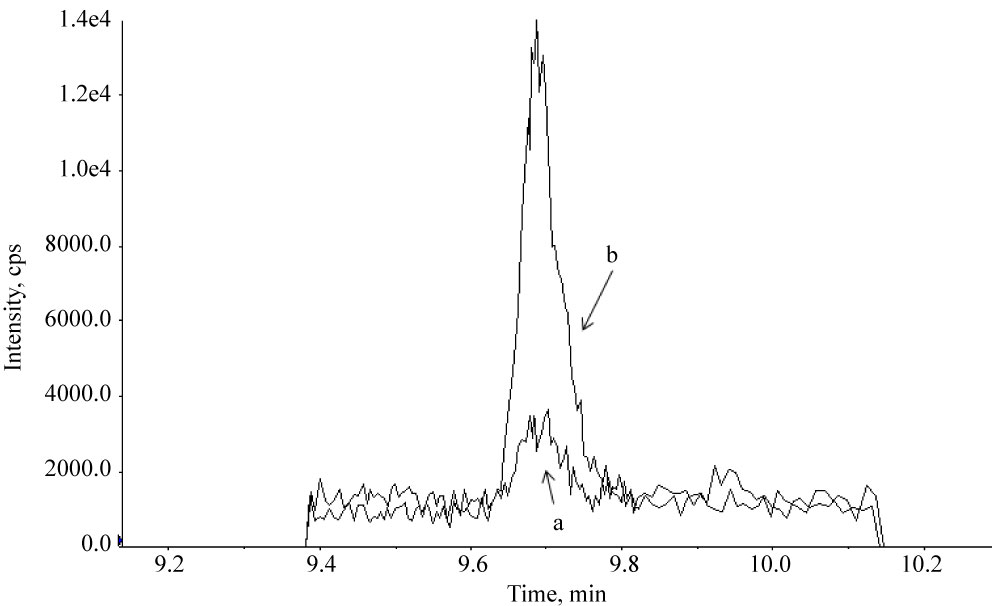
Supplementary Figure 1: LC-MS/MS quantifier ion chromatograms...
LC-MS/MS quantifier ion chromatograms (sMRM (scheduled Multiple Reaction Monitoring) mode) of PBZ (Phenylbutazone) in a milk sample fortified at 0.5 RC (recommended concentration): a) Without addition of ascorbic acid; b) with addition of ascorbic acid into milk samples. The extraction method was done with acetic acid.
The samples were prepared on the same day and analyzed during the same LC-MS/MS sequence. It can be clearly seen that the absence of ascorbic acid leads to the poor intensity.
Tables
Table 1: Optimized ESI-MS/MS parameters for each of the studied analytes.
Table 2: Validation parameters and factors.
Table 3: Validation sets with the specified combinations of the factor levels.
Table 4: Validation results in milk.
Supplementary Table 1: CV (%) at MRL/RC (3 replicates) and r2 of the linearity plot evaluated in the experiment with formic acid.
References
- KE Leslie, CS Petersson-Wolfe (2012) Assessment and management of pain in dairy cows with clinical mastitis. Vet Clin North Am Food Anim Pract 28: 289-305.
- G Galati, S Tafazoli, O Sabzevari, TS Chan, PJ O’Brien (2002) Idiosyncratic NSAID drug induced oxidative stress. Chem Biol Interact 142: 25-41.
- A Whelton (1999) Nephrotoxicity of nonsteroidal anti-inflammatory drugs: Physiologic foundations and clinical implications. Am J Med 106: 13-24.
- European Commission (2001) Reference laboratory for residues of veterinary drugs. Workshop NSAIDs and validation according to SANCO 1805/2000, Berlin, Germany.
- (2009) Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. Off J Eur Union L15: 1-72.
- V Naidoo, MA Taggart, N Duncan, K Wolter, J Chipangura, et al. (2018) The use of toxicokinetics and exposure studies to show that carprofen in cattle tissue could lead to secondary toxicity and death in wild vultures. Chemosphere 190: 80-89.
- (2007) CRL guidance paper: CRLs view on state of the art analytical methods for national residue control plans.
- JA Turton, AC Havard, S Robinson, DE Holt, CM Andrews, et al. (2000) An assessment of chloramphenicol and thiamphenicol in the induction of aplastic anaemia in the BALB/c mouse. Food Chem Toxicol 38: 925-938.
- Commission Decision 2003/181/EC (2003) Commission Decision of 13 March 2003 amending Decision 2002/657/EC as regards the setting of minimum required performance limits (MRPLs) for certain residues in food of animal origin. Off J Eur Communities L 71: 17-18.
- EM Malone, G Dowling, CT Elliot, DG Kennedy, L Regan (2009) Development of a rapid, multiclass method for the confirmatory analysis of anti-inflammatory drugs in bovine milk using liquid chromatography tandem mass spectrometry. J Chromatogr A 1216: 8132-8140.
- A Gentili, F Caretti, S Bellante, L Mainero Rocca, R Curini, et al. (2012) Development and validation of two multiresidue liquid chromatography tandem mass spectrometry methods based on a versatile extraction procedure for isolating non-steroidal anti-inflammatory drugs from bovine milk and muscle tissue. Anal Bioanal Chem 404: 1375-1388.
- P Jedziniak, T Szprengier-Juszkiewicz, K Pietruk, E Śledzińska, J Żmudzki (2012) Determination of non-steroidal anti-inflammatory drugs and their metabolites in milk by liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem 403: 2955-2963.
- M Cronly, P Behan, B Foley, E Malone, S Martin, et al. (2010) Rapid multi-class multi-residue method for the confirmation of chloramphenicol and eleven nitroimidazoles in milk and honey by liquid chromatography tandem mass spectrometry. Food Addit Contam Part A 27: 1233-1246.
- E Dubreil-Chéneau, Y Pirotais, M Bessiral, B Roudaut, E Verdon (2011) Development and validation of a confirmatory method for the determination of 12 anti-inflammatory drugs in milk using liquid chromatography-tandem mass Spectrometry. J Chromatogr A 1218: 6292-6301.
- P Gallo, S Fabroccino, F Vinci, M Fiori, V Danese, et al. (2008) Confirmatory identification of sixteen non-steroidal anti-inflammatory drug residues in raw milk by liquid chromatography coupled with ion trap mass spectrometry. Rapid Commun Mass Spectrom 22: 841-854.
- T Peng, AL Zhu, YN Zhou, T Hu, ZF Yue, et al. (2013) Development of a simple method for simultaneous determination of nine subclasses of non-steroidal anti-inflammatory drugs in milk and dairy products by ultra-performance liquid chromatography with tandem mass spectrometry. J Chromatogr B 933: 15-23.
- Xie J, Peng T, Zhu A, He J, Chang Q, et al. (2015) Multi-residue analysis of veterinary drugs, pesticides and mycotoxins in dairy products by liquid chromatography-tandem mass spectrometry using low-temperature cleanup and solid phase extraction. J Chromatogr B 1002: 19-29.
- A Rúbies, L Guo, F Centrich, M Granados (2016) Analysis of non-steroidal anti-inflammatory drugs in milk using QuEChERS and liquid chromatography coupled to mass spectrometry: Triple quadrupole versus Q-Orbitrap mass analyzers. Anal Bioanal Chem 408: 5769-5778.
- E van Pamel, E Daeseleire (2015) A multiresidue liquid chromatographic/tandem mass spectrometric method for the detection and quantitation of 15 nonsteroidal anti-inflammatory drugs (NSAIDs) in bovine meat and milk. Anal Bioanal Chem 407: 4485-4494.
- Jian Wang, D Leung (2012) The challenges of developing a generic extraction procedure to analyze multi-class veterinary drug residues in milk and honey using ultra-high pressure liquid chromatography quadrupole time-of-flight mass spectrometry. Drug Test Anal 4: 103-111.
- Commission Decision 2002/657/EC (2002) Implementing council directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off J Eur Communities L221: 8-36.
- DA Bohm, CS Stachel, P Gowik (2011) Validated determination of eight antibiotic substance groups in cattle and pig muscle by HPLC/MS/MS. J AOAC Int 94: 407-419.
- BV Fisher (1984) Statistics in chemistry. Anal Proc 21: 443-448.
Author Details
Malka Britzi and Frieda Schwartsburd
Veterinary Services, Israel National Residue Control Laboratory, Kimron Veterinary Institute, Ministry of Agriculture and Rural Development, Israel
Corresponding author
Frieda Schwartsburd, Veterinary Services, Israel National Residue Control Laboratory, Kimron Veterinary Institute, Ministry of Agriculture and Rural Development, Derech HaMaccabim 62, Rishon LeTsiyon, Israel, Tel: 972-3-9688936.
Accepted: July 11, 2019 | Published Online: July 13, 2019
Citation: Britzi M, Schwartsburd F (2019) Development and Validation of a High-Throughput Method for the Determination of Eight Non- Steroidal Anti-Inflammatory Drugs and Chloramphenicol in Milk, using Liquid Chromatography-Tandem Mass Spectroscopy. Int J Analyt Bioanalyt Methods 1:005.
Copyright: © 2019 Britzi M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
A high-throughput LC-MS/MS method for simultaneous identification and quantitation of eight non-steroidal anti-inflammatory drugs (NSAIDs) and chloramphenicol (CAP) in bovine and ovine milk was developed. The method is capable of detecting and quantitating: Carprofen (CPF), tolfenamic acid (TLF), 5-hydroxy flunixin (FLU-OH), diclofenac (DCL), 4-methylaminoantipyrin (4MAA), meloxicam (MLX), ibuprofen (IBU), phenylbutazone (PBZ) and chloramphenicol (CAP) at their maximum residue limits (MRLs) or target analytical levels. Addition of ascorbic acid into the milk samples before the extraction step was found to be crucial for repeatability and intensity of PBZ and 4MAA. The method consists of the single extraction step with acetonitrile and a commercially available salt mixture, followed by evaporation of the supernatant and reconstitution. Complementary LC-MS/MS and LC-MS/MS/MS methods were developed for confirmation of ibuprofen and diclofenac respectively. The method was validated according to EU Commission Decision 2002/657/EC requirements. The method accuracy was in the range of 89-108% and coefficients of variation of the interday precision assessment varied between 3% and 16%. This method has been implemented at the Israel National Residue Control Laboratory for routine monitoring of NSAIDs and CAP residues in bovine and ovine milk.
Keywords
NSAIDs and CAP, Bovine and ovine milk, Method validation, LC-MS/MS
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are widely used both as human and veterinary medicines for their anti-inflammatory, analgesic and antipyretic properties. Studies have illustrated that NSAIDs are the second most prescribed class of veterinary drugs after antimicrobials and there is a severe concern that they can enter the human food chain [1]. Aside their beneficial therapeutic properties, NSAIDs have unwanted side effects in humans, such as aplastic anemia/agranulocytosis, gastrointestinal disorders [2] and changes in renal function [3]. Thus, the European Council (EC) recommends rigorous control of NSAIDs in food producing animals [4].
In the European Union (EU) a few NSAIDs are authorized for dairy cows with established maximum residues limits (MRLs) in milk: Tolfenamic acid (TLF) -50 µg/kg; flunixin (FLU) (marker residue 5-hydroxyflunixin) – 40 µg/kg; meloxicam (MLX) -15 µg/kg; metamizole (marker residue 4-methylaminoantipyrine (4MMA)) – 50 µg/kg; and diclofenac (DCL) with very low MRL of 0.1 µg/kg [5]. Carprofen (CPF) is licensed for use in dairy cattle and has no required MRL in bovine milk; the target value for the analytical method was set at 500 µg/kg for our in-house monitoring of the substance, due to occurrence of vultures' death from consuming livestock carcasses [6]. Ibuprofen (IBU) and phenylbutazone (PBZ) are non-authorized for veterinary use in food-producing animals and have no established MRLs in milk; IBU and PBZ should be analyzed (as defined in CRL Guidance Paper of 2007 [7]) at the recommended concentrations (RC) of: 10 µg/kg and 5 µg/kg, respectively.
Chloramphenicol (CAP) is a broad-spectrum antibiotic that has been banned for use in all food-producing animals by EU, USA and Canada because of its life-threatening side effects in humans. Common side effects in humans include bone marrow suppression, nausea and diarrhea [8]. A maximum required performance limit (MRPL) for CAP was set by Commission Decision 181/2003/EU at 0.3 µg/kg in all foods of animal origin, such as milk, fish, meat eggs, etc. [9].
The NSAIDs/CAP chemical structure diversity and the wide range of MRLs/RCs (from 500 µg/kg for CPF to 0.1 µg/kg for DCL) make the development of a NSAIDs/CAP multi-residue method a challenging task. Liquid chromatography (LC) coupled to tandem mass spectrometry (MS/MS) is the technique of choice for such multi-residue analysis in complex matrices such as milk. Acetonitrile has been utilized for NSAID and CAP extraction, effectively inducing milk protein precipitation [10-13]. Application of methanol [14], as well as acetonitrile/methanol [15] or acetonitrile/ethyl acetate [16,17] mixtures, as extraction solvents were also reported. In several methods, the extraction step includes addition of salts, such as ammonium acetate [12], sodium chloride [10,13] or ammonium acetate/magnesium sulfate [18] mixture. These salts induce separation of the milk and organic solvent mixture, affording extraction of the analytes from the matrix.
The methods proposed by Dubreil-Chéneau, et al. [14] and van Pamel and Daeseleire [19] do not include any cleanup steps after extraction. Other methods include extensive cleanup of the extracts, utilizing solid phase extraction (SPE) cartridges with different sorbents (C18, amino phases and polymeric), depending on the compounds included in the method [11,12,20]. QuEChERS cleanup (quick, easy, cheap, effective, rugged and safe) approach was also applied [18].
Herein we report the development and validation of a reliable and high-throughput multi-residue method for the routine analysis of eight NSAIDS (CPF, FLU-OH, MLX, PBZ, IBU, DCL, 4MAA, TLF) and CAP in milk. The presented method allowed validation of IBU monitoring in milk, which was not included in some previously reported methods [14,118,19]. Additionally, the method could be validated at the diclofenac MRL, which to the best of our knowledge was not achieved elsewhere [14].
Experimental
Materials and reagents
The analytical standards were all over 98% purity. Standards of 5-hydroxyflunixin (FLU-OH) and 4-methylaminoantipyrine (4MAA) were supplied by Clear Synthesis, India. Carprofen was purchased from Dr. Ehrenstorfer, Germany; tolfenamic acid (TLF) and chloramphenicol (CAP) were obtained from Toronto Research Cemicals, Canada; diclofenac (DCL) from Sigma, Israel; ibuprofen (IBU) from Alfa Aesar, USA; meloxicam (MLX) and phenylbutazone (PBZ) were purchased from A2S, France; chloramphenicol-d5 (CAP-d5) and flunixin-d3 (FLU-d3) from Witega, Germany.
Methanol LC-MS grade was obtained from Merck, Israel; formic acid Optima LC/MS was from Fisher Scientific, UK. Acetic acid glacial, methanol, dimethyl sulfoxide and acetonitrile AR grade were purchased from Bio-Lab, Israel. L-ascorbic acid (99.9%) was obtained from Merck, Israel. Formic acid 98-100% was supplied by J.T. Baker, USA. MicroPure system, Thermo Electron LED, Germany, was utilized for ultrapure water (resistance > 18 mΩ). Anhydrous extraction salts mixes MgSO4/NaCl (4 gr/1 gr) and MgSO4/NaAcetate (4 gr/1 gr) in Mylar pouches were supplied by United Chemical Technologies (UCT), USA.
Equipment
Vortex mixer (Ginie 2, Scientific Industries, USA), Laboratory centrifuges (Hermle Z 216 MK, Germany and Eppendorf 5810 R, Germany), Nitrogen evaporator (Zymark, USA), Micropipettes (Finnpipette, Thermo Scientific, USA).
Standard solutions
Stock solutions of each analyte were prepared at a concentration of 1 mg/ml in different solvents as follows: carprofen, ibuprofen, tolfenamic acid and diclofenac were prepared in methanol/acetonitrile (1:9 v/v) solution; meloxicam, 5-hydroxyflunixin, phenylbutazone, flunixin-d3, and 4-methylaminoantipyrine were prepared in methanol/dimethyl sulfoxide (1:1 v/v) mixture; chloramphenicol and chloramphenicol-d5 were prepared in acetonitrile. Intermediate single standard solutions of FLU-d3, DCL, CAP and CAP-d5 were prepared in methanol at a concentration of 10 µg/ml. Intermediate single standard solutions of IBU and PBZ were prepared in methanol at concentrations of 500 and 250 µg/ml, respectively. A standard NSAIDs' fortification solution in methanol included CPF, FLU-OH, TLF, MLX, 4MAA, IBU, PBZ, and DCL at the concentrations of 50, 4, 5, 1.5, 5, 1, 0.5, and 0.01 µg/ml, respectively. A single standard fortification solution of CAP in methanol was prepared at the concentration of 0.1 µg/ml. An internal standards (IS) fortification solution of CAP-d5 and FLU-d3 was prepared in methanol at the concentrations of 0.1 µg/ml and 1 µg/ml, respectively. All standard solutions, excluding NSAIDs' fortification solution, were stored at +4 ℃ up to 6 months. The NSAIDs' standard fortification mix was prepared monthly. Fresh ascorbic acid solution (100 mg/ml) was prepared in purified water before each experiment.
Sample preparation
Bovine and ovine raw milk samples were utilized in the current study. An aliquot of 10 ml of milk was placed into a 50 ml polypropylene centrifuge tube, and 0.4 ml of ascorbic acid (100 mg/ml) was added into the tube. Samples were fortified with IS solution (50 µl) at levels corresponding to 5 ng/ml for FLU-d3 and 0.5 ng/ml for CAP-d5. After fortification, the samples were vortex-mixed, and 1 ml of glacial acetic acid was added to each sample followed by vortex mixing. Acetonitrile (10 ml) was added to each tube followed by vortex mixing. Then the entire content of extraction salts pouch MgSO4/NaCl (4 gr/1 gr) was added to each sample, which was immediately and vigorously vortex mixed, in order to disperse salts. The samples were centrifuged (3900 rpm, 15 min, 20 ℃).
An aliquot of 4 ml from the upper acetonitrile layer was transferred into a clean glass culture tube and evaporated under nitrogen at 60 ℃ to dryness. The dried samples were reconstituted in 0.4 ml of methanol/water solution (1:1, v/v), transferred into 1.5 ml micro-centrifuge tubes and subsequently centrifuged (13500 rpm, 10 min, 20 ℃). The extracts in the micro-centrifuge tubes were left overnight at + 4 ℃. On the next day the samples were additionally centrifuged (13500 rpm, 10 min, 20 ℃) and transferred into HPLC glass vials for LC-MS/MS analysis.
LC-MS/MS analysis
The LC-MS/MS system consists of a liquid chromatograph NexeraX2 (Shimadzu, Japan) coupled to a triple quadrupole mass spectrometer QTRAP 6500 (AB SCIEX, Canada) controlled by Analyst 1.6.3 software. Data analysis was performed using Multiquant 3.0 software. Kinetex XB-C18 column (2.6 µm, 100 Å 100 × 2.1 mm), Phenomenex, USA was used to separate NSAIDs and CAP. The column temperature was maintained at 40 ℃. The injection volume was 2 µl.
The main LC-MS/MS method: gradient consisting of 0.1% formic acid in water (mobile phase A) and methanol (mobile phase B) at a flow of 0.4 ml/min was utilized. The initial gradient conditions were 10% of mobile phase B, and then mobile phase B was linearly increased during 12 minutes up to 95%, and brought back to 10% between 12 and 12.5 min. The system was allowed to re-equilibrate at the initial mobile phase composition for 3 minutes. The total run time was 15.5 minutes.
The analysis was performed using electrospray ionization (ESI) operated in positive -negative polarity switch, the data were acquired utilizing scheduled multiple reaction monitoring (sMRM), with MRM detection window: 45 sec and target scan time: 0.3 sec. Source conditions were as follows: temperature: 350 ℃; capillary voltage: 5 kV/-4.5 kV; curtain gas: 35 psi; collision activated dissociation (CAD) gas: medium; nebulizer gas: 60 psi; turbo gas: 40 psi. Two product ions (quantifier and qualifier) were monitored for each analyte. MS/MS parameters and chromatographic retention times are summarized in Table 1.
IBU confirmation method: IBU confirmation was performed by MRM-IDA-EPI (information dependent acquisition- enhanced product ion) acquisition method. In the LC method the injection volume was set to 5 µl, in order to increase the sensitivity, while the other parameters were remained the same as in the main method. The EPI method utilized negative polarity with collision energy: 35 V, collision energy spread: 15 V and CAD gas: high, the ESI parameters remained the same as described above.
DCL confirmation method: DCL confirmation in the samples was achieved by a separate LC-MS/MS/MS method. In the LC method the injection volume was set to 20 µl, for obtaining DCL MS/MS/MS chromatograms in samples spiked at 0.5 MRL, while other parameters were remained the same as in the main method. The MS/MS/MS method operated in positive polarity, CAD gas - high, ESI parameters remained as mentioned above. First precursor (parent ion) was 296 Da, second (first generation product ion): 250 Da, DP: 50 V, collision energy: 30 V, collision energy spread: 15 V.
Method validation
The validation procedure meets the requirements of the 2002/657/EC guidelines [21,22]. This procedure permits choosing important factors that can influence the results of the analytical method and varying these factors on at least two levels. Subsequently the method is validated for the changes in these factors and the performance characteristics are calculated.
The presented study was designed and evaluated, considering changes in the factors that may occur during routine analysis. The following factors were chosen to be varied: milk type, temperature of evaporation, timing of the LC-MS/MS analysis and operator. The factors within the proposed study were varied on two levels (indicated as + or -), as described in Table 2. It was found that 50 assays should permit a satisfactory validation if conducted as follows: Two calibration curves (four concentration levels and a blank, 10 samples), and 40 validation samples (8 blanks and 32 spiked samples). The study allowed evaluating the following method validation parameters: linearity, specificity, accuracy (trueness), repeatability, interday precision, ruggedness, decision limit (CCα) and detection capability (CCβ).
Raw bovine and ovine milk pools containing non detectable levels of the analytes were utilized as blank samples in the experiment. The pools were prepared by mixing samples, received from various dairy farms across the country, for routine analysis.
Eight sets of five samples (validation samples, VS) were carried through the validation procedure, where each set reflected a specified combination of factor levels, as shown in Table 3. Each set included blank and four samples fortified at 0.5, 1, 1.5, and 2 MRL/RC of each analyte. Since there were no available certified reference materials for the analytes and the matrix of interest, an additional set of blank and four spiked samples (0.5, 1, 1.5 and 2 MRL/RC) of each milk type (bovine and ovine) was used as calibration samples (CS) for building matrix-matched calibration curves. The calibration samples were carried through the validation procedure with the specific combination of factors levels (two operators, two milk types, and temperature of evaporation 60 ℃, direct LC-MS/MS analysis). Two calibration curves were built by plotting the response factors (analyte peak area/ IS peak area) as a function of analytes concentrations in calibration samples. The levels of the analytes in the ovine and bovine validation samples were determined relative to the respective calibration curve.
The specificity of the method was verified by analysis of the 10 blank samples. The guidance for the specificity acceptance criteria was as follows: a response factor of interference in a blank sample at the retention time of interest had to be less than 30% of the LOQ response factor of the corresponding analyte. The LOQ value for each analyte was established as the lowest level on a matrix matched calibration curve that complies with criteria of accuracy and precision. In the presented study the LOQ level of each analyte was defined as the corresponding 0.5 MRL/RC.
The interday precision (2 days, 4 replicates) for each fortification level was evaluated as follows: The mean concentration at each fortification level (8 replicates) in validation samples was calculated, subsequently standard deviation (SD) and coefficient of variation (CV, %) were determined for each spiking level. Accuracy (%) was assessed by dividing the aforementioned mean concentration in the VS (8 replicates) by the theoretical fortification value: accuracy (%) = 100 × Xmean/Xtheo. Linearity was determined by plotting the mean of the calculated concentrations in the all validation samples at each spiking level as a function of the theoretical spiking levels. The ruggedness of the method was assessed by comparing the calculated MRL/RC values of each factor level for each parameter according to the Youden approach. The extensive explanation of the method was described earlier [21,23]. CCα and CCβ were calculated according to the procedure described in the Commission Decision 2002/657/EC [21].
Results and Discussion
Development of LC-MS/MS conditions
Method development was initiated by optimizing MS/MS conditions utilizing direct infusion of analytes, each at the concentration of 100 ng/ml, in methanol/(0.1% formic acid) (1:1, v/v) solution. Two most intensive transitions of the precursor ion were selected as quantifier and qualifier peaks to yield 4 identification points (1 for parent ion, 1.5 for each daughter ion) as required by CD 2002/657/EC [21].
A number of C18 chromatographic columns by Phenomenex were assessed in this study: Synergy Fusion-RP (4 µ, 80 Å, 50 × 2 mm), Kinetex XB-C18 (2.6 µm, 100 Å, 100 × 2.1 mm) and Kinetex C18 (2.6 µm, 100 Å, 100 × 2.1 mm). Kinetex XB-C18 (2.6 µm, 100 Å, 100 × 2.1 mm) demonstrated the optimal chromatographic performance, utilizing methanol/(0.1% formic acid) gradient at 0.4 ml/min flow rate with a total run time of 15.5 minutes, including the equilibration interval. Figure 1 demonstrates quantifier transitions chromatograms of extracted milk samples fortified with studied analytes at 0.5 MRL/RC.
The scheduled multiple reaction monitoring (sMRM) technique was utilized for improving sensitivity of the analytes with low validation concentration levels: CAP, IBU, DCL and PBZ. It should be noted that the main LC-MS/MS method failed to detect the reproducible qualifier fragments of IBU and DCL in the milk extracts. In the case of IBU this could be attributed to low abundance of the qualifier ion. The qualifier fragment of DCL was overlapped by some interference from the matrix that could not be separated chromatographically.
Two complementary LC-MS methods were developed for confirmation of the presence of IBU and DCL in milk samples as described in the Experimental section, Figure 2. The reproducible qualifier fragment of IBU was observed in the milk samples allowing confirmation of the IBU by the fragment ratio. Figure 3 demonstrates the sMRM scan chromatograms of IBU quantifier and qualifier ions of the extracted milk sample spiked at 0.5 RC.
The MRM-IDA-EPI methodology did not deliver satisfactory results in the case of DCL in the milk samples - the second MRM transition was not consistent. Therefore, a LC-MS/MS/MS method was developed for confirmation of DCL in the milk extracts. MS/MS/MS conditions for DCL were optimized by infusing the standard into the mass spectrometer at the concentration of 100 ng/ml in methanol/(0.1% formic acid) (1:1, v/v) solution. The method produced a distinctive MS signal of DCL in the milk samples at the level as low as 0.5 MRL at the corresponding retention time.
Development of sample preparation method
The aim was to develop a simple and reliable extraction method (using commercially available reagents and salt mixes), suitable for a laboratory with high workloads. The evaluation of the repeatability and linearity during the method development was done by analyzing three replicates of blank and spiked samples (0.5, 1, 2 MRL/RC) in each trial.
Addition of ascorbic acid to milk samples before the extraction process proved to be a critical step, as was described earlier [14,18]. In our hands, extraction without ascorbic acid resulted in poor repeatability and response intensity of PBZ and MAA in milk samples (See Supplementary Figure 1). Thus, the required concenrtation of ascorbic acid was gradually optimized, and it was found that addition of 0.4 ml of ascorbic acid (100 mg/ml) to milk samples before spiking by the fortification solution resolves the issue.
Acetonitrile extraction of acidified milk samples (with formic acid or glacial acetic acid) followed by addition of salts mixes (MgSO4/NaCl (4 gr/1 gr) or MgSO4/NaAcetate (4 gr/1 gr)) was applied. Preliminary trials showed that using formic acid resulted in poor repeatability and linearity for all of the analytes (see Supplementary Table 1). Using glacial acetic acid led to satisfactory results, and milk samples were directly acidified by 1 ml aliquot of glacial acetic acid, followed by addition of acetonitrile. Then a salt mix was added, the sample was vortexed and centrifuged at 3900 rpm at 20 ℃.
An aliquot of the upper acetonitrile layer was evaporated to dryness, resulting in a thick residue at the bottom of the tube. The residue was reconstituted in methanol/water solution, and the cloudy reconstituted extract was centrifuged at 13500 rpm at 20 ℃, resulting in a clear supernatant. However, some further precipitation occurred in the samples, while waiting in the autosampler for analysis.
Filtration of the samples was omitted in order to avoid losses of the analytes on filters, as was reported earlier [10]. Instead, the reconstituted centrifuged extracts were left overnight at +4 ℃ in centrifuge micro-tubes, inducing further development of particulate matter in the extracts. Subsequently, the samples were centrifuged again at 13500 rpm at 20 ℃, and then the clear supernatant was transferred into glass HPLC vials for LC-MS/MS analysis.
This simple protocol works well only upon utilizing MgSO4/NaCl (4 gr/1 gr) salts mix (rather than MgSO4/NaAcetate (4 gr/1 gr) salts mix) at the extraction step. Following the established protocol, the extracts in vials remained clear even after five days at +15 ℃. Using MgSO4/NaAcetate (4 gr/1 gr) salt mix during the extraction step resulted in the presence of floating particulate matter in centrifuged samples even after overnight at +4 ℃.
Method validation
The criteria for relative retention times and ion ratios were examined for all the samples and standards in the validation study. The values for these were in agreement with the EU requirements for all the analytes investigated in this study. In terms of relative retention time, the analyte peaks in samples were found to be within 2.5% tolerance as compared with the standards. Two transition ions were monitored for the eight of nine analytes. The most intense ion was used for quantitation. Ion ratios of eight analytes in the samples were within the required tolerances, as required by EU criteria, when compared with standards used during the validation study. Confirmation of the ninth analyte - DCL in the milk samples was achieved by utilizing the LC-MS/MS/MS method. The presented method passed the specificity acceptance criteria for all of the analytes.
Table 4 demonstrates the assessed accuracy values for each analyte at the respective 0.5, 1, 2 MRL/RC. The validation results show high accuracy for all of the analytes in the range of 89-108%. The method demonstrated low values (3-16%) of interday precision, expressed as the coefficient of variation (CV (%)) values at 0.5, 1, 2 MRL/RC, as described in Table 4.
The linearity of the chromatographic response was demonstrated by plotting the mean value of the calculated concentrations in VS (8 replicates) as a function of the theoretical values. All r2 values lay between 0.93 and 0.99 (see Table 4). The ruggedness test demonstrated that there was no significant effect of the varied parameters (Table 3) on the results of the analysis. CCα and CCβ values for each of the nine analytes are shown in Table 4.
Conclusions
An LC-MS/MS multi-residue method for determination of an antibiotic - CAP and 8 NSAIDs - CPF, DCL, TLF, FLU-OH, PBZ, MLX, IBU and 4MAA was developed and validated for bovine and ovine milk. The performance characteristics of the method in terms of linearity, repeatability, specificity, accuracy, ruggedness and interday precision met the requirements of the 2002/657/EC guidelines. This simple method is intended for control laboratories, allowing high sample throughput. The presented method has been successfully applied in routine analysis of milk samples at the Israel National Residue Control Laboratory.