
International Journal of Experimental Spectroscopic Techniques
(ISSN: 2631-505X)
Volume 3, Issue 2
Research Article
DOI: 10.35840/2631-505X/8519

Article Formats
Principles of High-Resolution NMR Spectroscopy of Paramagnetic Molecules in Solutions
Table of Content
Figures

Figure 1: A) The NMR spectra range of...
A) The NMR spectra range of cholesterol in CCl4; В) the NMR spectra of the same substance with the addition of Eu(DPM)3·2Py.
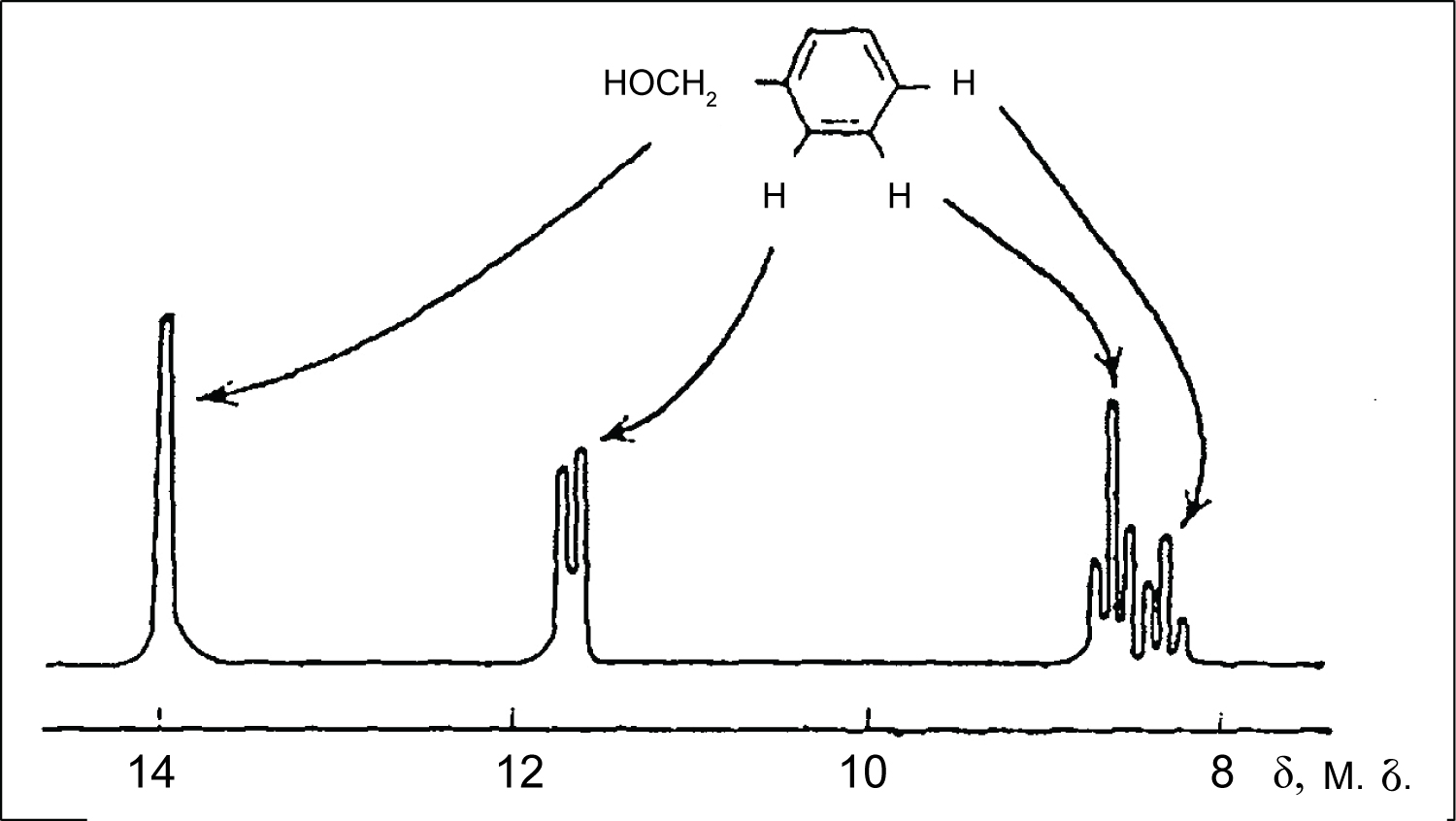
Figure 2: NMR spectrum of benzyl alcohol...
NMR spectrum of benzyl alcohol in the presence of Eu(DPM)3.
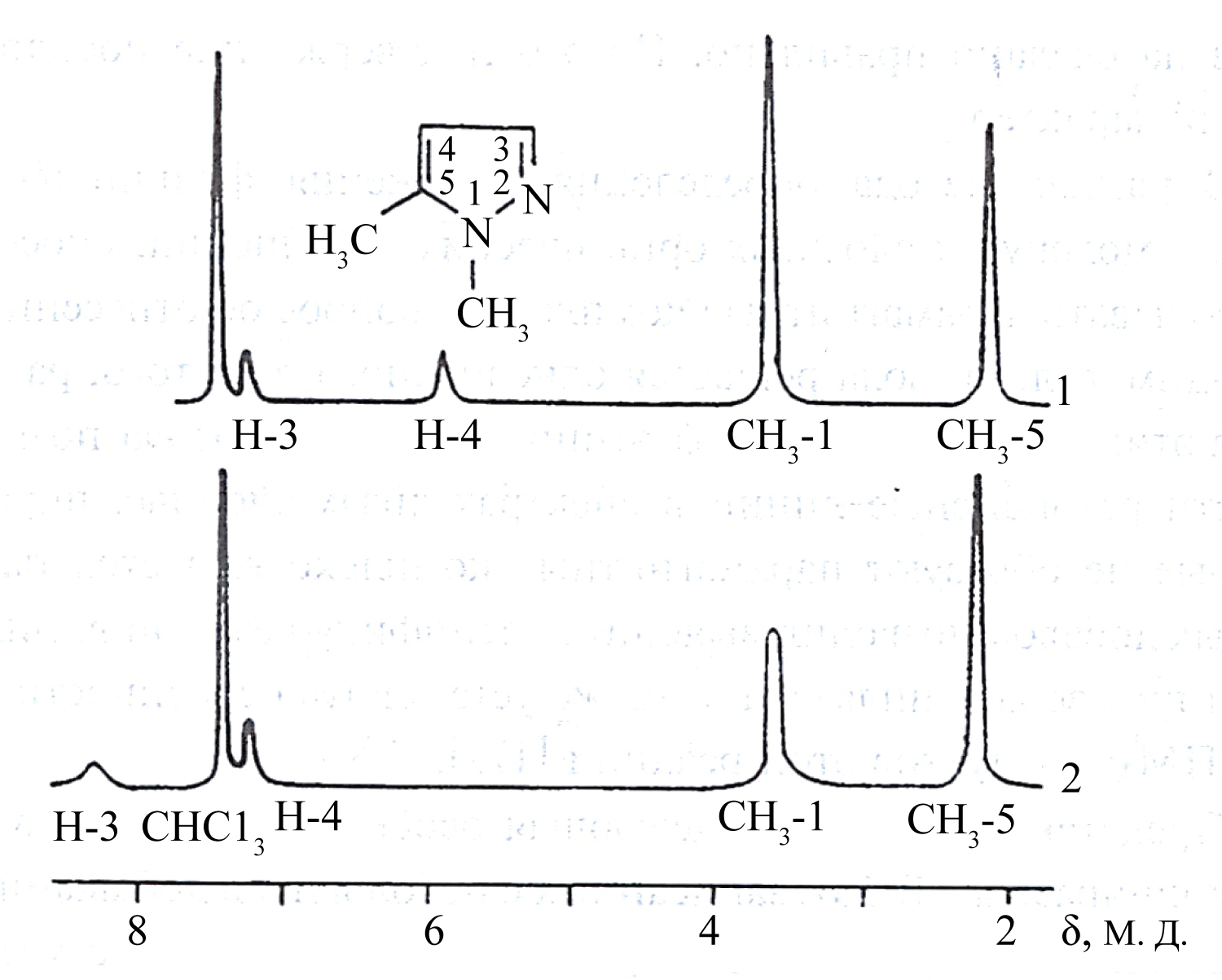
Figure 3: NMR spectrum of 1,5-dimethylpyrazole...
NMR spectrum of 1,5-dimethylpyrazole without (1) and with (2) additive of NiBr2·3H2O.

References
- Alkorta I, Elguero J, Denisov GS (2008) A review with comprehensive data on experimental indirect scalar NMR spin-spin coupling constants across hydrogen bonds. Magn Reson Chem 46: 599-624.
- Lodewyk MW, Siebert MR, Tantillo DJ (2012) Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem Rev 112: 1839-1862.
- Babailov SP (2008) Lanthanide paramagnetic probes for NMR spectroscopic studies of molecular conformational dynamics: Applications to macrocyclic molecules. Progress in Nuclear Magnetic Resonance Spectroscopy 52: 1-21.
- Voronov VK, Ushakov IA (2010) High resolution nuclear magnetic resonance in paramagnetic complexes. Russian Chemical Reviews 79: 835-847.
- Di Pietro S, Piano S, Di Bari L (2011) Pseudocontact shifts in lanthanide complexes with variable crystal field parameters. Coordination Chemistry Reviews 255: 2810-2820.
- Bertini I, Luchinat C, Parigi G (2011) Moving the frontiers in solution and solid state bioNMR. Coordination Chemistry Reviews 255: 649-663.
- Eaton DR, Phillips WD (1968) Nuclear magnetic resonance in paramagnetic compounds. Zh Strukt Khim 9: 153-183.
- Hinckley CC (1969) Paramagnetic shifts in solutions of cholesterol and the dipyridine adduct of trisdipivalomethanatoeuropium(III). A shift reagent. J Am Chem Soc 91: 5160-5162.
- Sanders JKM, Williams DH (1970) A shift reagent for use in NMR spectroscopy. A first-order spectrum of n-hexanol. Chem Com 7: 422-423.
- Voronov VK, Podoplelov AV (2015) Paramagnetic complexes in high resolution NMR spectroscopy. NOVA publishers, New York, USA, 181.
- Zaev EE, Voronov VK, Shwartsberg MS, Vasilevsky SF, Molin Yu N, et al. (1968) Application of paramagnetic additions to the structure determination of some pyrazoles by NMR. Tetrahedron Letters 5: 617-622.
- Andersson J, Hedin P, Johansson J, Nordström I, Nydén M (2010) Coordination of imidazoles by Cu(II) and Zn(II) as studied by NMR relaxometry, EPR, far-FTIR vibrational spectroscopy and ab initio calculations: Effect of methyl substitution. J Phys Chem A 114: 13146-13153.
- Jaroniec CP (2012) Solid-state nuclear magnetic resonance structural studies of proteins using paramagnetic probes. Solid State Nucl Magn Reson 43-44: 1-13.
- Voronov VK, Ushakov IA, Shmelev VV, Sagdeev DR (2012) Peculiarities of intramolecular exchange and valence tautomerism in metal semiquinolates determined by high-resolution NMR spectroscopy. Magn Reson Chem 50: 350-356.
- Voronov VK (2016) NMR spectra transformed by electron-nuclear coupling as indicator of structural peculiarities of magnetically active molecular systems. J Phys Chem A 120: 6688-6692.
- Babailov SP, Peresypkinaa EV, Journaux Y, Vostrikova KE (2017) Nickel(II) complex of a biradical: Structure, magnetic properties, high NMR temperature sensitivity and moderately fast molecular dynamics. Sensors and Actuators B: Chemical 239: 405-412.
- Abraham RJ, Filippi M, Petrillo G, Piaggio P, Vladiskovic C, et al. (2017) A theoretical and NMR lanthanide‐induced shift (LIS) investigation of the conformations of lactams. Magn Reson Chem 55: 1059-1072.
- Voronov VK, Ushakov IA, Baikalova LV (2005) NMR spectra of 1-vinylimidazole paramagnetic complexes with of iron group elements. Russian Chemical Bulletin 54: 1473-1476.
Author Details
VK Voronov*
Irkutsk National Research Technical University, Irkutsk, Russia
Corresponding author
VK Voronov, Irkutsk National Research Technical University, Irkutsk, Russia.
Accepted: October 11, 2018 | Published Online: October 13, 2018
Citation: Voronov VK (2018) Principles of High-Resolution NMR Spectroscopy of Paramagnetic Molecules in Solutions. Int J Exp Spectroscopic Tech 3:019.
Copyright: © 2018 Voronov VK. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The review summarizes the peculiarities of NMR phenomenon in paramagnetic molecules. These peculiarities are due to hyperfine or electronic coupling between the unpaired electrons and the resonant nuclei that leads to the paramagnetic broadening as well as to the paramagnetic shifts (contact and pseudocontact) of resonance lines in NMR spectra. The contact shift is observed when a probability of the unpaired electron disposition in the location of resonance nucleus is different from zero. Therefore, these shifts can be considered as a source of information about nature of the metal-ligand bond as well as about electron structure of the ligands. Pseudocontact shifts characterize the spatial structure of the molecule and can be used for solving various structural problems. The review covers the pioneering works describing the specifics of the NMR spectra transformed by additives of paramagnetic complexes of iron group elements including those of cobalt and nickel as well as complexes of rare-earth elements, e.g. europium.
Keywords
NMR spectroscopy, Hyperfine coupling, Paramagnetic shifts
Introduction
Just after the discovery of nuclear magnetic resonance (NMR), this phenomenon has formed the basis of a powerful method for structural study of compounds and their properties, at least, in liquid phase. For a long time, diamagnetic compounds remained the main objects of the structural investigations using NMR technique (see, for example, ref. [1,2] and the references cited therein). Despite the fact that this situation is still retained, there are strong grounds to believe that study of the NMR phenomenon in paramagnetic systems will also allow obtaining valuable information on molecular structure of the compounds. Such opportunity relates, as a rule, to paramagnetic complexes, which specifics are defined by unpaired electrons on d- and f- orbitals. Electron-nuclear or hyperfine coupling (HFC) between unpaired electrons and nuclei of the paramagnetic molecule leads to characteristic shifts and broadenings associated with spatial and electronic structure of the molecules, which NMR spectrum is detected, by explicit functional dependence. In paramagnetic compounds, diverse relaxation effects can be observed that essentially expands research possibilities of the NMR method. The investigations of temperature dependence of paramagnetic shifts and signal broadenings enable to establish thermodynamic parameters of complex formation in solution and to elucidate stoichiometry of complexes and peculiarities of intra- and intermolecular exchange processes. Hence it follows that the NMR spectra, transformed by HFC, permit, at least, in principle, a considerable body of unique information on molecular structure to be obtained [3-6].
General NMR Phenomenon in Paramagnetic Molecules
The specifics of NMR phenomenon in paramagnetic systems is defined by a character of hyperfine coupling between unpaired electrons and resonance nuclei that leads to broadening and shifts (contact and pseudo-contact ones) of the resonance lines. The contact shift is observed when a probability of the unpaired electron disposition in the location of resonance nucleus is different from zero. For a paramagnetic complex with insignificant spin-orbital coupling, the value of contact shift ωk is defined by equation of Bloembergen-McConnel [7]:
Where A is a constant of contact hyperfing coupling determined by Fermi formula; γe and γN - are gyromagnetic ratio for electron and nucleus of N; ge is g factor of electron; S - is a spin of a complex; β - is Bore magneton; Т - is absolute temperature; k - is Boltzmann constant.
Therefore, these shifts can be considered as a source of information about nature of the metal-ligand bond as well as about electron structure of the ligands. The excellent examples illustrating efficiency of the contact HFC application for structural study of multielectron systems are given in the review [7].
The pseudocontact shift is observed when the dipole-dipole coupling between magnetic moments of the unpaired electrons and nuclei is not averaged to zero. Implicitly, this shift is defined as follows:
Here terms f(g) and f(φ,r) are determined by magnetic anisotropy and geometry of the complex, respectively. Explicit form of these functional dependencies and, hence, value and sign of ωp depend on symmetry of the paramagnetic complex and nature of the coordinating ion.
The pseudo-contact shifts characterize spatial structure of the molecule that explains their use for the solution of different structural problems. The most striking example of successful application of pseudo-contact shifts for these purposes is a method of lanthanide shifting reagents offered in the works [8,9].
It is reported [8] on comparability of the NMR spectra of cholesterol solution recorded without and with additives of the paramagnetic Eu(DPM)3∙2Py complex. The use of the latter is explained by very short time of electron relaxation (τ) of rare-earth element ions. Therefore, the complex formation leads to insignificant broadening of resonance lines, whereas times of their paramagnetic shifts are considerable. Figure 1 shows the NMR spectra of cholesterol in a solution of carbon tetrachloride without (A) and with (B) additive of Eu(DPM)3·2Py [8]. In the spectrum of the free ligand (cholesterol) protons of the metal as well as the methylene and methine groups, not associated with the oxygen atom, resonate in a strong field, forming an unresolved multiplet. The remaining protons resonate in a weaker field. Coordination of cholesterol with europium ion leads to markedly offset resonance lines (spectrum B).
This allows eventually holding the most complete classification of signals. The ability of Eu(DPM)3∙2Py to induce relatively high paramagnetic shifts, which depend upon concentration of the complex in the studied sample without essential broadening of resonance lines, allows to conclude that this complex can be used as a shifting reagent.
After Hinckley has published his works, Sanders and Williams also reported that Eu(DPM)3 is an excellent shifting reagent [9]. Being coordinated with lone electron pair of the functional group, this europium compound induces considerable paramagnetic shifts. For example, in the PMR spectra of benzylic alcohol, aromatic protons are responsible for a broad singlet. After addition of Eu(DPM)3 to a solution, the spectrum becomes available to the analysis according to the rules for spectra of the first order. The induced shifts quickly decrease with the increase in number of the bonds between hydroxyl group and the corresponding protons. When Eu(DPM)3 is added to a solution of N-hexanol, spectrum of the latter can also be analyzed according to the rules for spectra of the first order (Figure 2) [9].
The works [8,9] have initiated a large number of publications (especially in the first half of the 70th of the twentieth century), including reviews (more than 40), appearing sometimes simultaneously in various countries. A number of them remain relevant to the present time. Perhaps some of them are now only of historical interest. However, it was the pioneer work that laid the foundations for the successes in using paramagnetic complexes for spectral analysis, which made so far [10].
In addition to the above publications [8,9] it is appropriate to mention the work [11], the authors of which, apparently, for the first time, used a paramagnetic broadening resulting from HFC, in structural studies. Figure 3 shows the spectra of an isomer of dimethylpyrazole recorded without and with the additive of NiBr2·3H2O.

Scheme: Remote location of the resonance nuclei from the coordination ion.
Since the broadening of Н4 proton signal, which position in the spectrum is known, is significantly lesser than that of a signal of other pyrazole proton, this isomer represents 1,5-dimethylpyrazole, where r5-6 < < r3-6 (r - distance from the coordinating ion to the atom nucleus, which signal is detected; see next section), that illustrates the diagram below (scheme). Before publication of the work [11], there were no reliable methods for identification of pyrazole isomers formed by hydrogen substitutions in the positions five and three.
Moreover, interpretation of the spectra of 3-and 5-substituted pyrazole allows including resonance lines in the spectra of disubstituted pyrazoles, which do not form paramagnetic complexes. As shown by the authors of this work, a method for decryption of the NMR spectra of complex organic molecules, based on the use of NMR characteristics in paramagnetic systems, is sufficiently reliable and simple.
As a rule, one fails in recording the high-resolution NMR spectra of paramagnetic complexes because of significant broadening of the signals. This relates, first of all, to the NMR signals from carbon, nitrogen and other atoms which constitute skeleton of the molecule. For example, signals broadening from protons can reach hundreds and even thousands hertz. The carbon signals can broaden so significantly that their detection becomes impossible at all. The values of paramagnetic shifts can be quite high (in comparison with the typical spectral range of diamagnetic compounds). Besides, the paramagnetic shifts can be different in sign that complicates detecting of signals related to individual paramagnetic particles (complexes and radicals). However, it turns out that these difficulties can be circumvented if to employ samples containing considerable excess of ligands as compared to concentration of the coordination ion. Then the spectrum becomes average and depends, in particular, upon character of hyperfine coupling in a paramagnetic complex. For a long time (up to the beginning of the ninetieths of the last century), overwhelming number of works dedicated to usage of HFC specifics for studying molecular structure were carried out in such a way.
The increase of a number of compounds, both synthesized and isolated from natural feedstock, significantly expands a gamut of products appropriate for HFC studies. Also, the situation began to change with the appearance of commercial Furrier-spectrometers working in a pulse mode. The application of modern NMR spectrometers allows recording the spectra of paramagnetic systems in a wide spectral range (about 400 ppm for 1H NMR and 2000 ppm for 13C NMR). Ultra-speed work stations of such spectrometers are capable of processing a huge number of the NMR spectra that makes it possible (at least, in principle) to assign the resonant signals. The usage of temperature attachments enables to perform complex dynamic investigations involving paramagnetic systems. Therefore, we are sure that studies of paramagnetic compounds using high resolution NMR spectroscopy will be progressing intensively. Efficiency of HFC application for the study of the molecular structure using modern pulsed NMR spectrometers has been illustrated, for example, in the works [3,5,8,12-17].
Method of Paramagnetic Additives
As it has already been noted, very often it is impossible to record the high-resolution NMR spectrum of the paramagnetic complex due to considerable broadening of the resonance lines.
This especially relates to the NMR resonance signals from carbon, nitrogen and some other atoms, which constitute skeleton of the molecules.
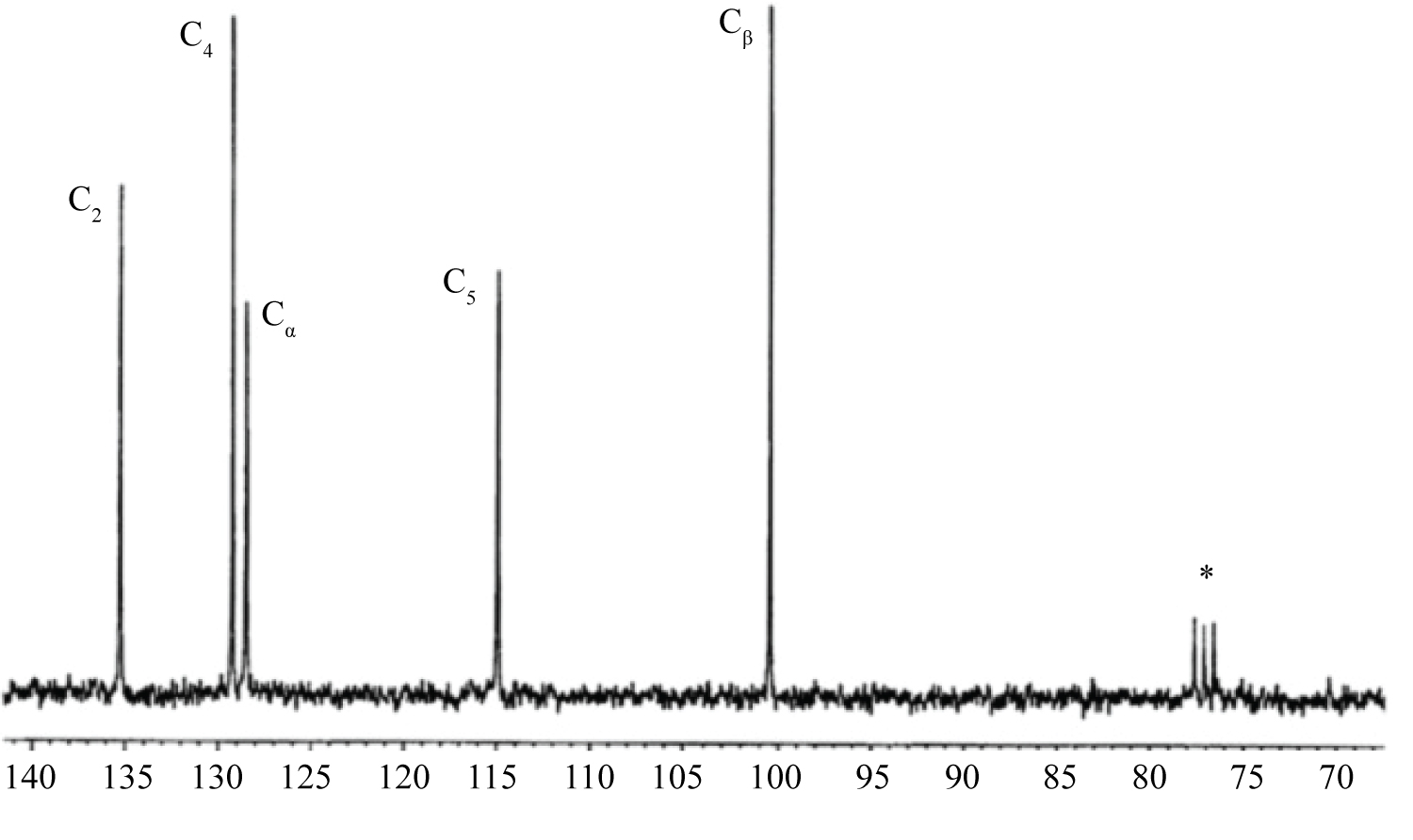
For example, broadenings of signals from protons can reach hundreds and even thousands hertz. As for the signals of carbon atoms, they can broaden so significantly that their detection becomes problematic. Figure 4 and Figure 5 depict the 13C NMR spectra of 1-vinylimidazole (VIM) and its complex with СоCl2, (VIM)4·СоCl2, recorded at room temperature (CDCl3 as a solvent). The spectrum of VIM is recorded with proton decoupling; the spectrum of VIM-cobalt complex is recorded without proton decoupling [18]. Here and in other figures, the solvent signals are designated by the asterisk. As is expected, the spectrum shown in Figure 5 contains five signals. The signal of the vinyl carbon atom C(α) is present as a doublet owing to spin-spin coupling with Н(Х) proton, while signal of the С(β) atom appears as a triplet due to spin-spin coupling with Н(А) and Н(В) protons. Carbon atoms of the imidazole cycle cause the emergence of three strongly broaden signals, which are also significantly shifted relative to their positions in the 13C NMR spectrum of non-coordinated VIM (see Figure 4). In the spectrum of VIM-nickel complex, only signals of the vinyl group atoms (i.e. С(α) and С(β)) are observed, whereas signals of carbon atoms of the imidazole ring are not detected owing to their strong paramagnetic broadening. For this reason, in the 13C NMR spectrum of the copper complex, the signals from carbon atoms are not detected at all.
The aforementioned spectral peculiarities are explained by the fact that the relaxation times of electron spins in complexes of cobalt, nickel and copper are significantly different. The cobalt complex has the shortest relaxation time (approximately 10-12 s), while for the copper complex, this value is considerably higher (~10-9 s). The shorter the relaxation time, the shorter will be the contact of resonating nuclei with uncompensated electron spins. Eventually, transformation of the NMR spectrum will be less expressed, and this is observed in the spectra discussed here. It should be added that among the elements of iron subgroup, the paramagnetic ion Со2+ can be used as a shifting reagent. In some cases, Ni2+ ions may also be suitable for this purpose.
The difficulties caused by paramagnetic broadening can be circumvented if to record the spectra of samples, in which the metal - ligand ratio (Р) is much less than 1. Due to the fast-molecular exchange between the coordination sphere of the complex and free ligand, the observed spectrum becomes average. Therefore, it would depend, in particular, on the nature of electron-nuclear coupling in paramagnetic complex, as well as on its spatial structure (the relative location of ligand molecules and coordinating ion). In case of rapid exchange, the observed broadening (Δ) of signals in the NMR spectrum is defined by [4]:
Where δ - is the observed paramagnetic resonance shift spin line (signal offset this resonated in the spectrum kernel with paramagnetic additives against position in the spectrum of a sample that does not contain paramagnetic additive); r - is a distance from the coordination ion to resonating nucleus; B and C - are constants of the studied sample at given temperature. Thus, for the complexes, in the spectra of which paramagnetic shifts are pseudo-contact, the broadenings of signals are defined by the expression:
The paramagnetic additives method comprises the recording of NMR spectra with or without additives of a paramagnetic complex. Molecules of solvent should not coordinate with paramagnetic additive. This is the main requirement, which is imposed to solvents. When the lanthanide shifting reagents are employed, the typical solvents are CCl4, CHCl3, and benzene. Quite often CH3OH is used as a solvent, if compounds, coordinated with lanthanide shifting reagents by pyridine nitrogen atom, act as a substrate. Deuterated analogs of these solvents now became readily available. If elements of the iron group are employed as paramagnetic additives, the suitable solvents are (apart from CHCl3) CH3NO2 and DMSO. HMDS is used as internal standard.
Usual temperature for recording of the NMR spectra is 45 ÷ 50 ℃, if lanthanide shifting reagents are employed as additives. For iron additives, the temperature can be significantly higher: it depends on realization of the fast exchange conditions. Identical broadenings of all signals in the NMR spectra irrespective of the distance between resonating nucleus and coordination ion evidence the absence of the above conditions [4]. Concentration P of paramagnetic additive relative to the substrate dissolved in the sample should be 10-3 - 10-2. In this range, one should prepare 7 - 10 additives. This is sufficient, first, to be convinced in linear character of δ = δ(P)dependence of the observed shifts. As our experience shows, to obtain qualitative 1H spectra, it is sufficient to dissolve about 10 mg of substrate (with molecular weight of 100 - 200) in a sample having standard volume (0.7 cm3). Such amount of substance is sufficient to quickly record qualitative spectra (with a good signal/noise ratio). Certainly, the modern spectrometers, operating in the pulse mode, allow to work (in principle) almost with any quantity of the substrate. Here we mean concentrations, which make it possible to obtain the relevant spectral information during small periods of time.