
International Journal of Metallurgy and Metal Physics
(ISSN: 2631-5076)
Volume 3, Issue 1
Research Article
DOI: 10.35840/2631-5076/9211

Article Formats
Thermodynamics and Elastic Properties of Precipitates with Orthorhombic, Hexagonal and Cubic in Al-Li-Cu-Mg Alloys
Table of Content
Figures
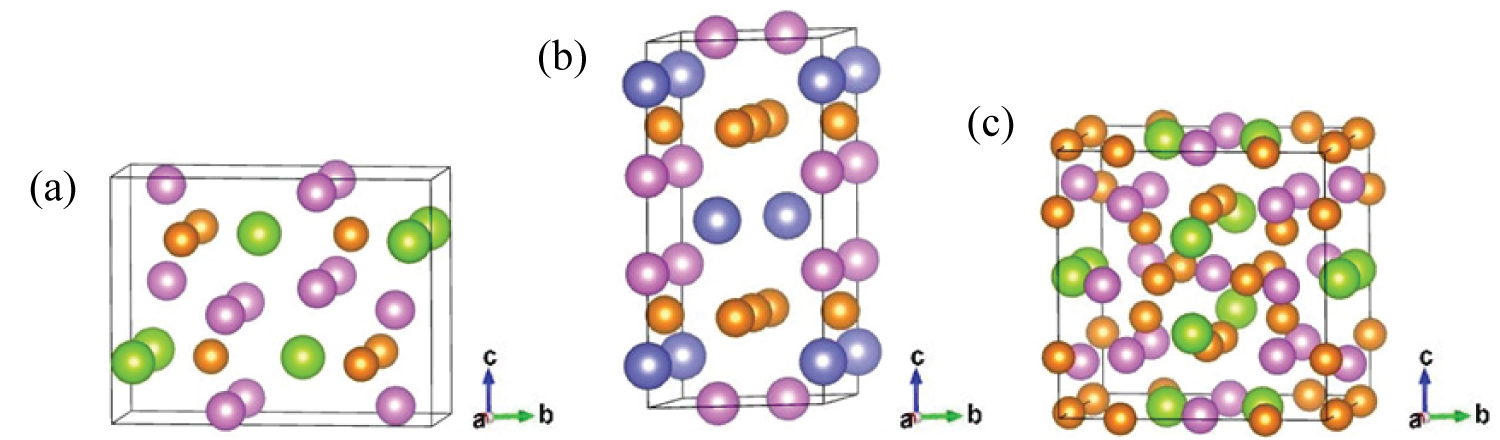
Figure 1: The crystal models of S (a) T1 (b) and σ...
The crystal models of S (a) T1 (b) and σ (c). The pink, yellow, green and purple ball denote Al, Cu, Mg and Li atom, respectively.
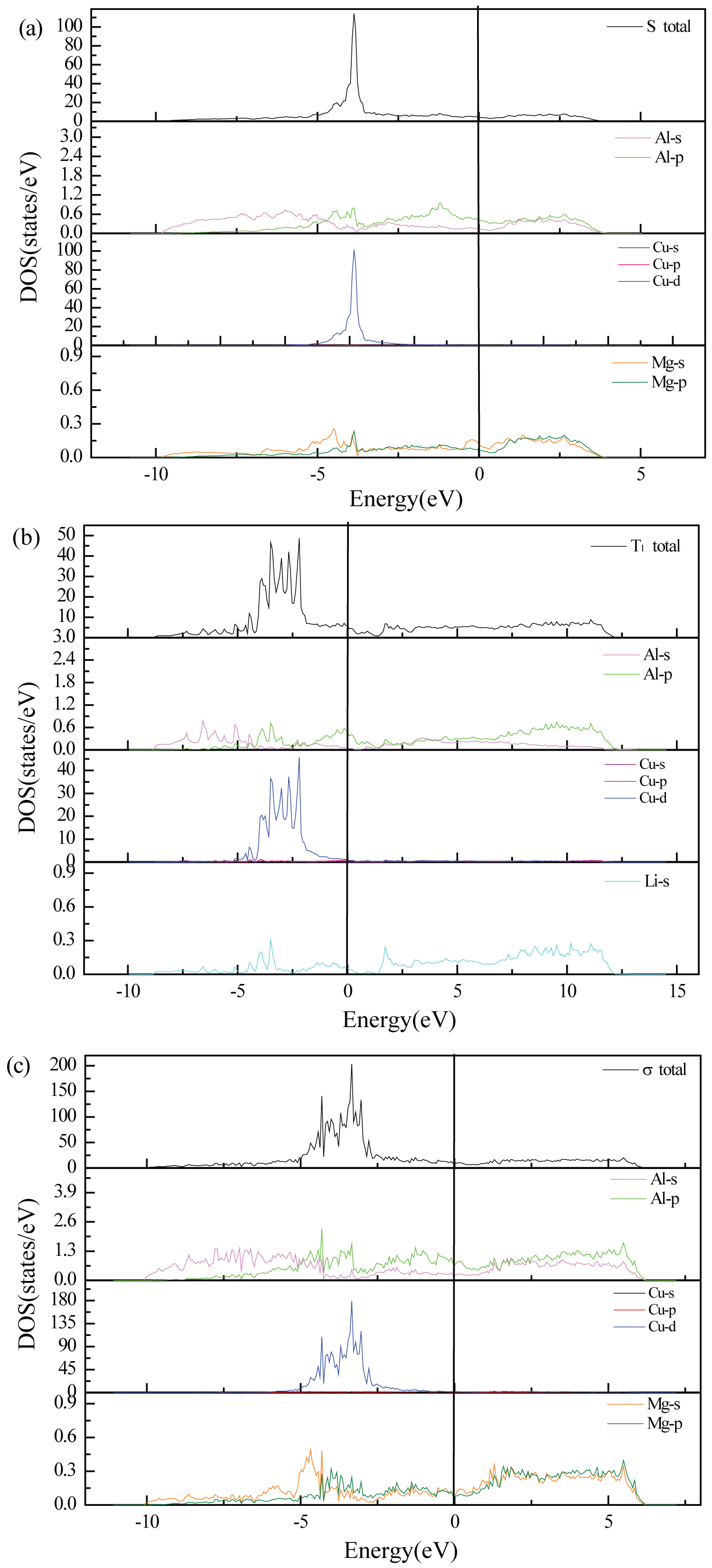
Figure 2: Total and partial density of states of S, T1 and σ...
Total and partial density of states of S, T1 and σ a) S; b) T1; c) σ. The vertical solid lines indicate the location of Fermi level.
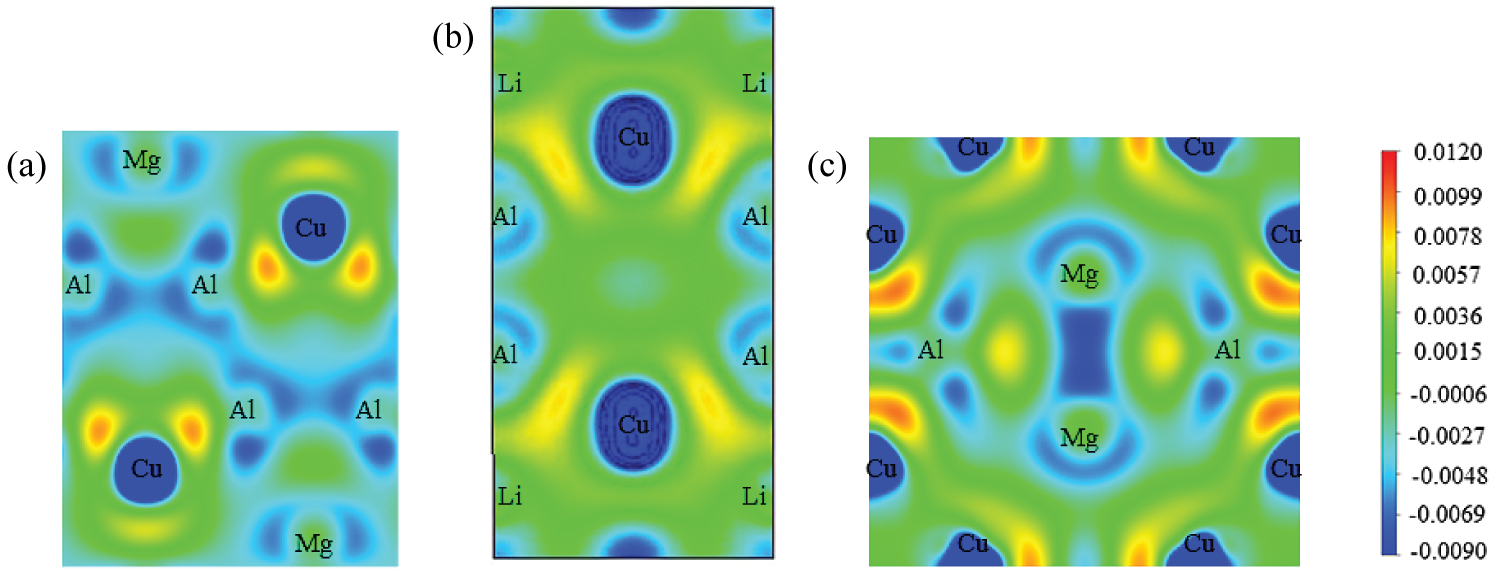
Figure 3: Distribution of charge density...
Distribution of charge density difference for S (a) T1 (b) and σ (c) along (100) plane.
Tables
Table 1: Calculated and experimental lattice constant, cohesive energy and heat of formation of S, T1 and σ.
Table 2: The relationship between elastic strain energy and strain of S, T1 and σ.
Table 3: The calculated and experimental elastic constants (GPa), bulk modulus (GPa), shear modulus (GPa), Young' modulus (GPa), and Possion's ratio of S, T1, σ and Al.
Table 4: The Debye temperature of S, T1 and σ.
References
- RK Gupta, Niraj Nayan, G Nagasireesha, SC Sharma (2006) Development and characterization of Al-Li. Materials Science and Engineering A 420: 228-234.
- JD Kim, JK Park (1993) Effect of stretching on the precipitation kinetics of an A1-2.0Li-2.8Cu-0.5Mg (0.13Zr) alloy. Metallurgical and Materials Transactions A 24: 2613-2621.
- YJ Gu, A Wahab, Z Huang, YG Zhang, CQ Chen (2001) The structure transformation in an Al-Li-Zn-Mg-Cu-Zr alloy. Materials Science and Engineering A 316: 39-45.
- ML Bairwa, PP Date (2004) Effect of heat treatment on the tensile properties of Al-Li alloys. Journal of Materials Processing Technology 153-154: 603-607.
- CM Li, SM Zeng, ZQ Chen, NP Cheng, TX Chen (2014) First-principles calculations of elastic and thermodynamic properties of the four main intermetallic phases in Al-Zn-Mg-Cu alloys. Computational Materials Science 93: 210-220.
- J Padilla, D Vanderbilt (1997) Ab initio study of BaTiO3 surfaces. Physical Review B 56: 1625-1631.
- JA White, DM Bird (1994) Implementation of gradient-corrected exchange-correlation potentials in Car-Parrinello total-energy calculations. Phys Rev B Condens Matter 50: 4954-4957.
- MD Segall, PLD Lindan, MJ Probert, CJ Pickard, PJ Hasnip, et al. (2002) First-Principles Simulation: Ideas, Illustrations and the CASTEP Code. J Phys Condens Matter 14: 2717-2744.
- K Laasonen, A Pasquarello, R Car, C Lee, D Vanderbilt (1993) Car-Parrinello molecular dynamics with Vanderbilt ultra soft pseudopotentials. Phys Rev B Condens Matter 47: 10142-10153.
- Y Jin, C Li, M Yan (1990) On the crystal structure of S'phase in Al-Cu-Mg alloy. Journal of Materials Science Letters 9: 421-424.
- JC Huang, AJ Ardell (2013) Crystal structure and stability of T1, precipitates in Al-Li-Cu alloys. Materials Science and Technology 3: 176-188.
- S Samson (1949) The crystal structure of Mg2Cu6Al5. Acta Chem Scand 3: 835-843.
- JP Perdew, K Burke, M Emzerhof (1996) Generalized gradient approximation made simple. Phys Rev Lett 77: 3865-3868.
- LP Huang, ZQ Zheng, YP Huang (2004) Microstructure and properties of 2197 Al-Li alloy. The Chinese Journal of Nonferrous Metals 14: 2066-2071.
- Z Gao, JH Chen, JZ Liu, ZR Liu, CL Wu (2012) Study on the evolution law of T1 phase in Al-Cu-Li alloy. Journal of Chinese Electron Microscopy Society 31: 308-314.
- BR Sahu (1997) Electronic structure and bonding of ultralight LiMg. Materials Science & Engineering: B 49: 74-78.
- Hideo Nakae, Hidetoshi Fujii, Katsuya Nakajima, Akiko Goto (1997) Infiltration and combustion synthesis of an intermetallic compound Ni3A1. Materials Science and Engineering: A 223: 21-28.
- ZQ Wen, YH Zhao, H Hou, PD Han (2014) First-principles study on doping effect of 5d transition metal in Ni3Al. Rare Metal Materials and Engineering 6: 1381-1386.
- WC Hu, Y Liu, DJ Li, XQ Zeng, CS Xu (2013) Mechanical and thermodynamic properties of Al3Sc and Al3Li precipitates in Al-Li-Sc alloys from first-principles calculations. Physica B Condensed Matter 427: 85-90.
- SQ Wang, HQ Ye (2003) Ab initio elastic constants for the lonsdaleite phases of C, Si and Ge. Journal of Physics: Condensed Matter 15: 5307-5314.
- P Ravindran, L Fast, PA Korzhavyi, B Johansson, J Wills, et al. (1998) Density functional theory for calculation of elastic properties of orthorhomcic crystals: Applications to TiSi. Journal of Applied Physics 84: 4891-4904.
- AH King (2013) Diffusion induced grain boundary migration. Metallurgical Reviews 32: 173-189.
- S Prikhodko, Y Ma, A Ardell, D Isaak, J Carnes, et al. (2003) Elastic constants of face-centered cubic and L12 Ni-Si alloys: Composition and temperature dependence. Metallurgical and Materials Transactions A 34: 1863-1868.
- W Xu, J Han, Y Wang, C Wang, X Liu, et al. (2013) First-principles investigation of electronic, mechanical and thermodynamic properties of L12 ordered Co3(M, W) (M= Al, Ge, Ga) phases. Acta Materialia 61: 5437-5448.
- D Connétable, O Thomas (2011) First-principles study of nickel-silicides ordered phases. Journal of Alloys and Compounds 509: 2639-2644.
- JP Watt (1980) Hashin-Shtrikman bounds on the effective elastic moduli of polycrystals with monoclinic symmetry. Journal of Applied Physics 51: 5120.
- JP Watt, L Peselnick (1980) Clarification of the Hashin-Shtrikman bounds on the effective elastic moduli of polycrystals with hexagonal, trigonal, and tetragonal symmetries. Journal of Applied Physics 51: 5125.
- J Hanies, JM Léger, G Bocquillon (2001) Synthesis and Design of Superhard Materials. Annual Review of Materials Research 31: 1-23.
- Y Lu, D Jia, F Gao, Z Chen, T Hu (2014) First-principles study on the elastic properties of Sr-Ti-O ceramics. Solid State Commun 182: 43-46.
- P Dawson, D Boyce, SM Ewen, Ronald Rogge (2001) On the influence of crystal elastic moduli on computed lattice strains in AA-5182 following plastic straining. Materials Science and Engineering A 313: 123-144.
- ZX Song (2010) First-principle calculations of precipitation phase in Al-Cu-Mg alloys. Hunan University.
- DQ Sun, YX Wang, XY Zhang, MY Zhang, YF Niu (2016) First-principles calculation on the thermodynamic and elastic properties of precipitations in Al-Cu alloys. Superlattices and Microstructures 100: 112-119.
- L Fu, JL Ke, Q Zhang, BY Tang, LM Peng, et al. (2012) Mechanical properties of L12, type Al3X (X = Mg, Sc, Zr) from first-principles study. Physica Status Solidi 249: 1510-1516.
Author Details
Yixin Zhang, Yongxin Wang*, Zheng Chen and Dongqiang Sun
Department of Materials Science, Northwestern Polytechnical University, China
Corresponding author
Yongxin Wang, State Key Laboratory of Solidification Processing, School of Materials, Northwestern Polytechnical University, Xi'an 710072, China, Tel: +8613384900383.
Accepted: March 20, 2018 | Published Online: March 22, 2018
Citation: Zhang Y, Wang Y, Chen Z, Sun D (2018) Thermodynamics and Elastic Properties of Precipitates with Orthorhombic, Hexagonal and Cubic in Al-Li-Cu-Mg Alloys. Int J Metall Met Phys 3:011.
Copyright: © 2018 Zhang Y, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
First-principles calculations based on density functional theory was used to investigate the structural, thermodynamic and elastic properties of ternary precipitations, S(Al2CuMg), T1(Al2CuLi) and σ(Al5Cu6Mg2), in Al-Li alloys. The results of lattice constants accord with experimental results well. The values of bulk modulus (B), shear modulus (G) and Young's modulus (E) can be a good reference in the absence of experimental data. The calculated data show that σ is the most thermodynamically stable and most trend-forming phase, which has the greatest effect in strengthening the matrix. Compared with S, T1 can improve the toughness of the matrix. The analysis of electronic structure suggests that three precipitations all exhibit metallic character, and bonding strength are the nature of different elastic properties for S, T1 and σ.
Keywords
First-principles, Aluminium alloys, Elastic properties, Alloy design
Introduction
Aluminum alloy has been the most widely used metal structural materials in the military industry, mainly used in aviation, aerospace and weapons industry. It can give full play to the potential of the material by making into various sections of the profiles, pipes, etc, as a result of its low density, high strength and good processing properties [1]. For each additional 1% of the lithium element in aluminum, its density can be reduced by 3%, the modulus increased by 5%. Therefore, the weight of the aircraft will be greatly reduced if the traditional aluminum alloy structural materials were replaced by aluminum-lithium alloy. The addition of Cu and Mg not only improved the strength of Al-Li alloy, but also changed the precipitation sequence of Al-Li alloy [2]. Precipitation strengthening is the main strengthening way of Al-Li alloy, so the nature of the precipitation is particularly important for the performance of the alloy [3]. The most common and important precipitations in Al-Li-Cu-Mg alloys are δ'(Al3Li), θ(Al2Cu), T1(Al2CuLi), S(Al2CuMg) and σ(Al5Cu6Mg2) [4,5]. Many researchers studied the morphology and evolution of T1 and S by electron microscopy, while the research on σ is very rare. The element Si added to the Al-Cu-Mg system alloys, which have the appropriate atom ratio of Cu to Mg would promote forming precipitating σ phase. Recent researches demonstrated that σ phase had good growth resistance property during 250 ℃, resulting in great room and high temperature performance. Thus, study of the σ phase precipitating from Al-Cu-Mg system alloys would give some experimental supports to the new Al-Cu-Mg alloys and improve their heat resistance, economic and comprehensive properties to meet the supersonic aircraft's demands. So far, there are no systematic reports on the mechanical and thermodynamic properties of the ternary precipitated in AI-Li-Cu-Mg alloys. In this work, first-principles method was performed to study the properties of Al-Li-Cu-Mg alloy precipitations including S, T1 and σ. Lattice constants, structural stability, elastic properties and electronic structure were discussed.
Calculation Method
All of the calculations, in this paper, were performed using the density function theory (DFT) as implemented in the Vienna Ab initio Simulation Package (VASP). The interaction between the ionic core and the valence electrons was described by Generalized gradient approximation (GGA) of the Perdew-Burke-Ernzerh of (PBE) functional [6,7]. The Brillouin zone is sampled using a Monkhorst-Pack k-point grid for S and σ, in addition, a Gamma centered is used for T1 of the hexagonal structure [8,9]. We considered the convergence of the results with respect to cutoff energy and k-points carefully. The cutoff energy for plane wave expansion was 450 eV for all the crystal models. The k-points meshes were set to be 18 × 8 × 10, 18 × 18 × 10 and 10 × 10 × 10 for S, T1 and σ, respectively. The valence electrons considered in their pseudopotential were 3s23p1 for Al, 3d104s1 for Cu, 2p63s2 for Mg and 2s1 for Li, respectively. The above parameters could assure a total energy convergence of 10-5 eV/atom and maximum force under 0.01 eV/Å.
Results and Discussion
Structural properties
The models of precipitations S, T1 and σ are shown in Figure 1, which are orthogonal [10], hexagonal [11] and cubic [12] structures, respectively. The lattice constants for S, T1 and σ were calculated from geometry optimization. Our calculated results, other theoretical and experimental results are listed in Table 1. Generally, the calculated results of the lattice constant and experimental value deviation within 5% can be regarded as credible [15]. It is obvious that our results are close to the experimental and other calculated values. The lattice constants calculated in this paper deviate from the experimental values by a maximum of 3%, which indicates that the calculated results are credible. Further, the calculation parameters used in this paper are credible.
Cohesive energy and heat of formation
Based on the optimization of geometrical structure, the cohesive energy and heat of formation of S, T1 and σ are calculated respectively as shown in Table 1. Table 1 also shows the corresponding experimental values and theoretical calculations for some of the precipitates. As for S and T1, it is found that the calculated cohesive energy and the formation heat are very close to the experimental values and the formation heat of S is closer to the experimental value than other theoretical values, which shows once again that our method is accurate and feasible. However, there is no experimental or theoretical value for σ. Therefore, our calculated results of σ can be a good reference for further study. The cohesive energy of a crystal is the energy released by the free atom binding to the crystal, that is, the work done when the intermetallic compound is decomposed into an isolated atom with infinite distance [16]. The strength and structural stability are closely related to the cohesive energy. The lower the value of cohesive energy, the more stable the crystal formed. The cohesive energy can be calculated by the following equation [16]:
Where Etot is the total energy of the precipitations in the equilibrium state; denote the energy of the isolated A, B and C atom, respectively; are the number of A, B and C atom in the primitive cell respectively. As we can see from Table 1, all the values of the cohesive energy are negative, which indicates the structures of S, T1 and σ phases are stable. The cohesive energy increases in the order of σ, S, and T1, so their corresponding stability decreases in turn. This shows that σ is the most stable phase and it will be the last one to disappear in the process of heating homogenization.
The formation of heat refers to the energy released by the formation of pure elements in the normal state, which can be used to characterize the intermetallic alloying ability of the compounds [17]. The lower the formation heat, the stronger the alloying ability of the phase, that is, the more easily formed during the formation of the alloy; otherwise, the more difficult to form [18]. Heat of formation can be calculated by using the formula as below [19]:
Where denotes the total energy of the precipitations in the equilibrium state,and are the energy of atom A, B and C in the solid states, respectively; are the number of A, B and C atom in the primitive cell, respectively. The values of heat of formation for S, T1 and σ are all negative, which reveals that S, T1 and σ are energetically favorable and have strong alloying ability. The highest value of the formation of heat is S, the lowest is σ and the middle is T1. It's meaning that σ has the largest tendency to form, followed by T1 and S, thermodynamically. In other words, there must be σ phase if you want to precipitate S or T1 in Al-Li-Cu-Mg alloy while satisfying the conditions of precipitation of the three phases. In combination with the formation of thermal values, we find that σ is the most stable and most easily formed phase. However, the cohesive energy of S is larger than that of T1, and the heat of formation is smaller than T1, indicating that S is more stable and the tendency of T1 is larger.
Elastic properties
The elastic constant () is an important parameter, which can reflect the mechanical properties of the material. The relationship between energy and strain that satisfies the Hooke's law under the condition of a small strain, and the elastic constant is the coefficient of this linear relationship. The elastic constant can be calculated by the following equation [20]:
Where is the volume; is the energy of the system; denote the strains applied in the and directions of the crystal, respectively.
The relationship between strain energy and strain can be obtained by calculating the strain energy of a single crystal under pressure in different directions. The elastic constant is the quadratic coefficient of the quadratic function, while the quadratic function is obtained by fitting the data of "energy change-strain amplitude". The relationship between strain energy and strain is [21,22]:
Due to the different symmetries, the way the strain is applied is different for different crystal systems. Table 2 shows the details of the strain applied to S, T1 and σ, as well as the relationship between elastic strain energy and strain.
The elastic constants calculated by the first-principles method are shown in Table 3. The values of elastic constants for S are in agreement with theoretical results. However, there is no experimental or theoretical value for T1 and σ. Therefore, our calculated results about elastic properties of T1 and σ can be a good reference for further study.
The mechanical stability of the crystal can be determined by the elastic constant.
For orthorhombic phase, the mechanical stability criteria are given by [24]:
For hexagonal phase, the mechanical stability criteria are given by [25]:
For cubic phase, the mechanical stability criteria are given by [26]:
According to the data in Table 3, it is found that the S with the orthorhombic structure, the T1 with the hexagonal structure and the σ with the cubic structure are all satisfy the conditions required by the equations (5) to (7), clearing that they are mechanically stable.
The bulk modulus () and the shear modulus () of the crystals corresponding to different crystal systems can be simplified as follows [27-29]:
Orthorhombic phase:
Hexagonal phase:
Cubic phase:
Young's modulus () and the Poisson's ratio () can be calculated using the following equations [30,31]:
The bulk modulus (), Shear modulus (), Young's modulus () and the Poisson's ratio () of S, T1, σ and Al can be calculated according to Equations (8) to (19). The results are shown in Table 3.
The values of the bulk modulus and shear modulus show that σ has the strongest deformation resistance and the strongest directional bonds between the atoms. It is displayed that S has the smallest bulk modulus, meaning that S has a weakest resistance to volume change by external stress. In other words, the precipitation of S phase will reduce the probability of cracks in the alloy, because the S phase will be deformed along with the matrix under the load conditions. Young's modulus [32] can represent a level of stiffness, and the larger the Young's modulus is, the stiffer the material is. The Young's modulus of the σ is 149.4 GPa, which is larger than that of the other two precipitations, arguing that the σ has the stiffest structure, followed by T1 and S. The shear modulus of each precipitated phase is much higher than that of the matrix, and the tendency is almost the same as the shear modulus. This phenomenon shows that these three precipitates can effectively obstruct the volume deformation of the alloy, especially the σ.
The ductility or brittleness is an important parameter for the alloys, which can be evaluated by the ratio of bulk modulus to shear modulus (B/G) and Poisson's ratio (υ). The greater the value is, the better the brittleness of the material is; conversely, the ductility is better, with a critical point of 0.57. The calculated results clear that T1 and σ are ductile materials and S is brittle material. The Poisson's ratio is another way to assess the ductile and brittleness of metals and its value is usually ranges from -1 to 0.5. The bigger the value of Poisson's ratio is, the better the ductility of material is. It shows that T1 has a best ductility, followed by σ and finally S, which is in accordance with the previous Pugh.
Debye temperature
The propagation speed of sound in a solid is the energy propagation velocity of the lattice vibration, and the specific thermal property as measured by the Debye temperature is caused by an increase in the vibrational level of the lattice. Therefore, The Debye temperature approximation can be obtained through the mechanical properties of the estimated sound velocity. The relationship between sound velocity and mechanical properties is [33]:
Where are the transverse phonon velocities, the longitudinal phonon velocities, and the average phonon velocities, respectively; is the density of the material; is the bulk modulus, and is the shear modulus. Debye temperature of the material can be obtained by phonon velocity [33]:
Where is the number of atoms; is the Avogadro's number; is Boltzmann's constant; is Planck's constant and is the relative molecular mass.
It can be seen from Table 4 that Debye temperature of the three precipitated phases is higher than that of the matrix, and the value of T1 is the largest, followed by S and σ, indicating that these precipitated phases will improve the Debye temperature of the alloy. The precipitation of T1 has the greatest contribution to the improvement of Debye temperature. Debye temperature is closely related to the specific heat and melting point properties of the material. It is also a measure of the close relationship between the atoms and can reflect the size of the expansion coefficient. Crystal will swell under high temperature conditions, and different phases will squeeze each other to produce cracks under different expansion coefficients, resulting in material failure. With its higher Debye temperature and lower coefficient of expansion, T1 becomes the least susceptible phase to high temperature cracking in the three major phases. The result is in agreement with the experiment.
Electronic structure
In order to investigate the nature of structural stability and chemical bonding, the total and partial density of state (DOS) of precipitations S, T1 and σ were calculated and plotted in Figure 2. The outer electronic structures of the isolated atoms involved in the calculations include Al 3s23p1, Cu 3d104s1, Mg 2p63s2 and Li 2s1. There is no pesudogap near the Fermi energy in the total density of states of the three precipitates, indicating that S, T1 and σ all have the metallic character. The three total DOS can be divided into two parts, the one shows a sharp peak, which is contributed mainly by the Cu 3d states, another part is dominated by Al 3s, Al 3p, Mg 2p, Mg 3s or Li 2s states.
In addition, the number of bonding electrons can be obtained from the total DOS, which is the average charge number of each cell from -10 eV to EF in this work. The larger number of bonding electrons is, the stronger charge interaction among bonding atoms is, and the more stable crystal structure is [5]. The values of number of bonding electrons for S, T1 and σ are 6.006, 4.139 and 6.506 respectively. The value of number of bonding electrons about S is close to σ, where σ is slightly better. It is obvious that the structural stability of T1 is weaker than that of σ and S. The results are in good accordance with the analysis of cohesive energy.
In order to get a more intuitive understanding of the bonding between atoms, the charge density difference of S, T1 and σ are also calculated. The calculated charge density difference along with (100) planes of S, T1 and σ were shown in Figure 3. There is a large amount of free electrons between Al-Al, Mg-Mg, Al-Mg, Al-Li, Cu-Li and Mg-Cu atoms, which illustrates that they all forming metal bonds. In addition, charge accumulation is obvious for the Al and Cu atoms of S, T1 and σ, and the charge density of σ is the highest, followed by S and T1. It means that the chemical bonds formed between Al and Cu atoms in σ is the strongest, which is consistent with the results of the elastic properties.
Conclusions
In summary, we investigated elastic and thermodynamic properties of three ternary precipitations, S, T1 and σ, in Al-Li-Cu-Mg alloys by first-principle method based on density functional theory (DFT). The central results are summarized as follows:
• Cohesive energy and heat of formation suggest that the structural stability of σ is best, followed by S and T1.
• Mechanical properties reveal that σ possesses the best reinforcement effect. And S is a brittle material, while T1 and σ are plastic materials.
• The total and partial state densities show that all of the three precipitates exhibit metallic properties, and the number of bonded electrons explains the stability of the precipitation in the Al-Li-Cu-Mg alloy. Charge density difference contributes to the differences of mechanical property for S, T1 and σ.
Acknowledgements
The authors would like to express their great gratitude for projects supported by the National Natural Science Foundation of China (Grant No.51475378, 51674205) and Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase).