A Novel Use of Oxidative Coupling Reactions Part II. Kinetic Spectrophotometric Method for Determination of Fluvastatin in Pharmaceutical Formulations

International Journal of Experimental Spectroscopic Techniques
(ISSN: 2631-505X)
Volume 8, Issue 1
Research Article
DOI: 10.35840/2631-505X/8534

Article Formats
A Novel Use of Oxidative Coupling Reactions Part II. Kinetic Spectrophotometric Method for Determination of Fluvastatin in Pharmaceutical Formulations
Table of Content
Figures
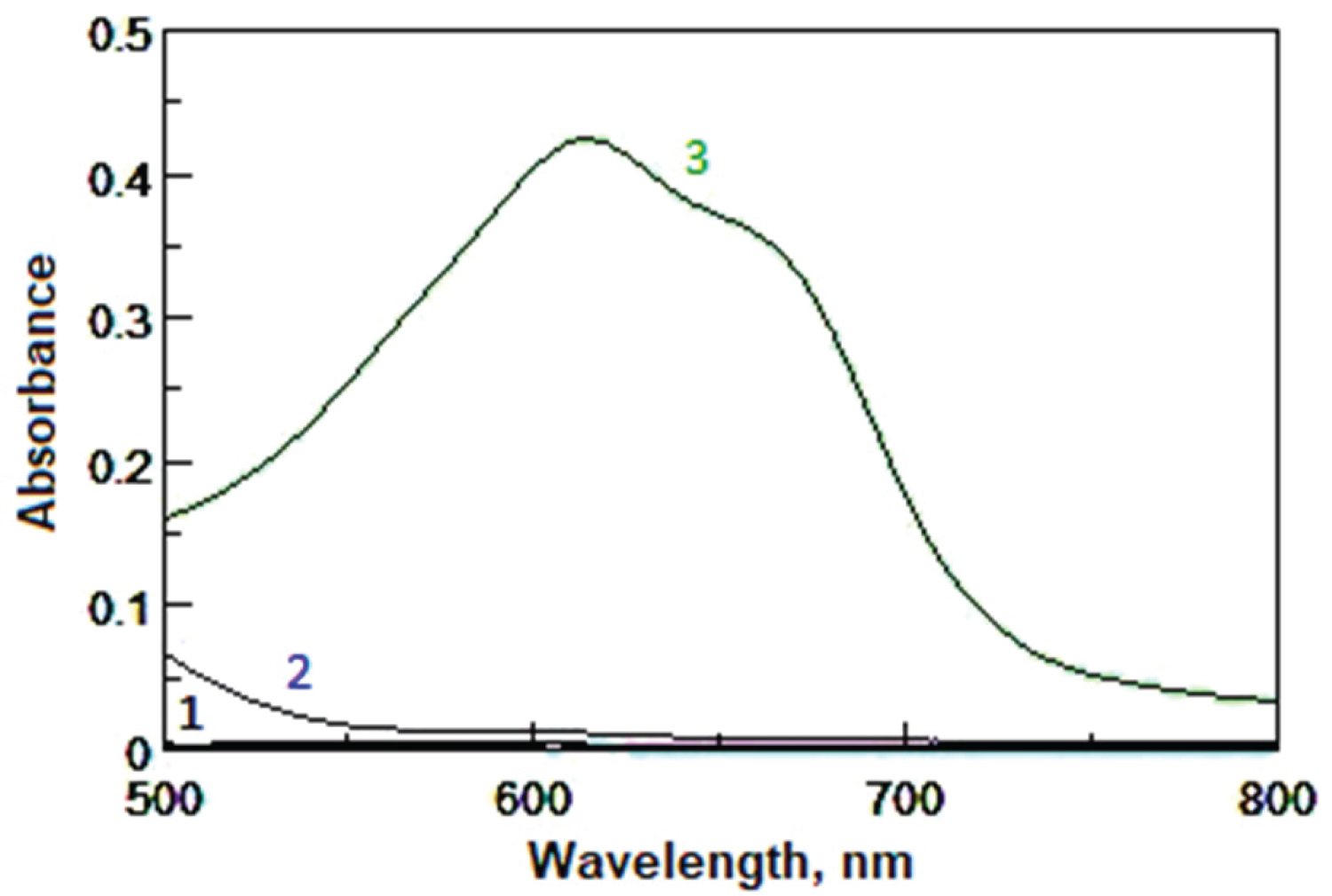
Figure 1: Absorption spectra of FVS....
Absorption spectra of FVS solution (20 µg/mL) against water (1), reagent blank against distilled water and (2) and FVS-MBTH system against reagent blank (3). CFVS = 20 µg/mL + 1.0 mL of MBTH 10-2 M + 1.0 mL of 1% Ce(SO4)2 in acid medium.
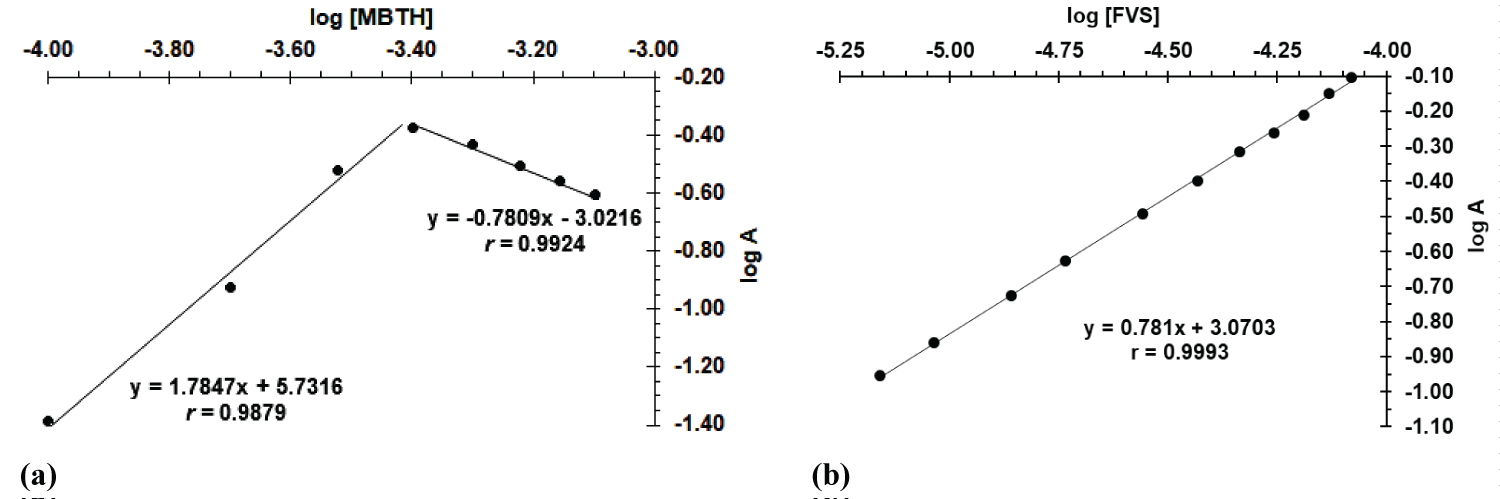
Figure 2: Limiting logarithmic plot for.....
Limiting logarithmic plot for stoichiometric ratio between FVS and MBTH. (a) logA vs. log[MBTH], CFVS = 4.6 × 10-5 M, CMBTH = 1.0 × 10-4 - 8.0 × 10-4 M and; (b) logA vs. log[FVS], CMBTH = 4.0 × 10-4 M, CFVS = 6.92 × 10-6 - 8.31 × 10-5 M.
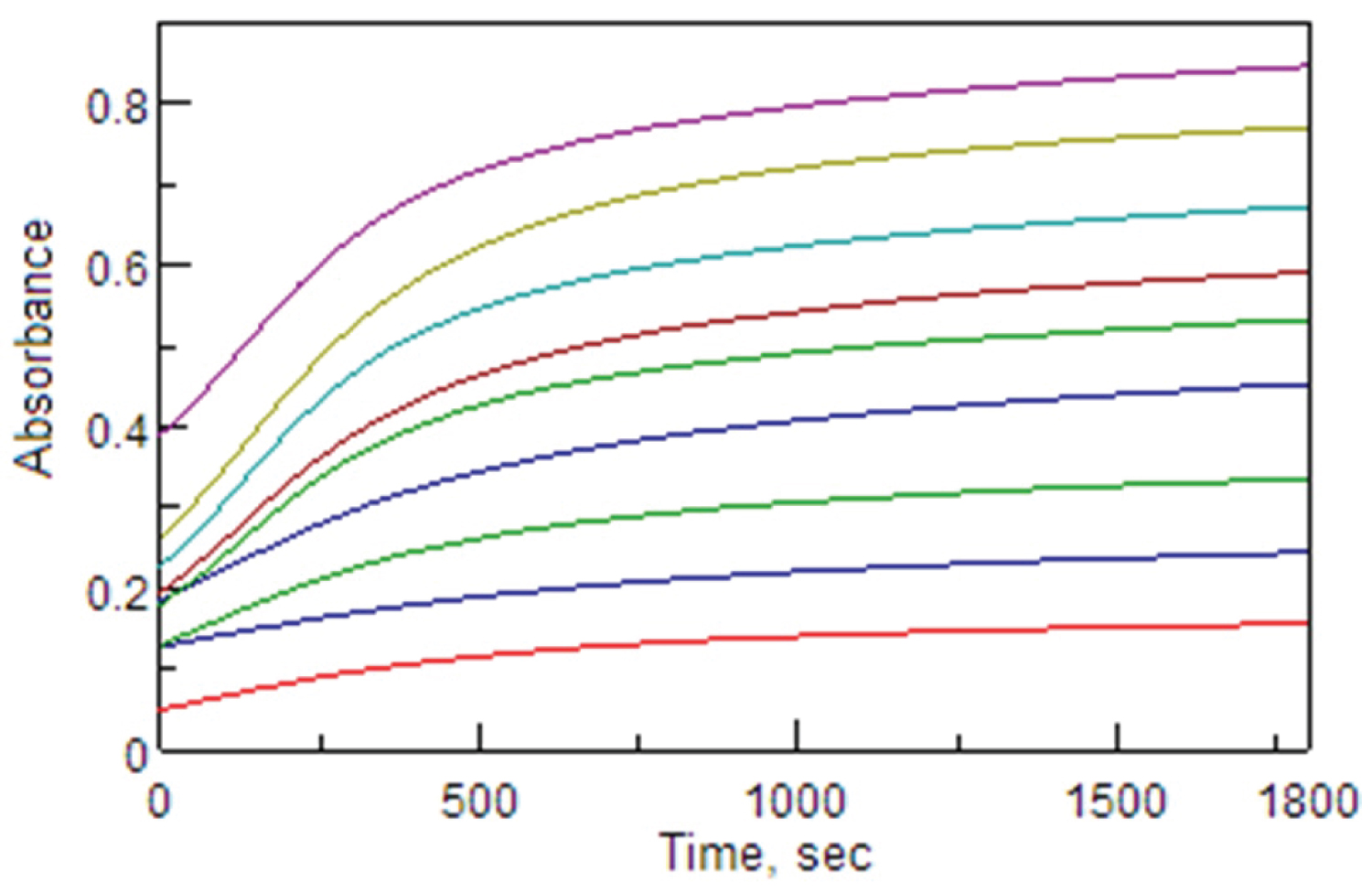
Figure 3: Absorbance-time curve for the....
Absorbance-time curve for the reaction of FVS with MBTH in aqueous acidic medium of Ce(SO4)2 at 615 nm; CFVS = 3-36 µg/mL.
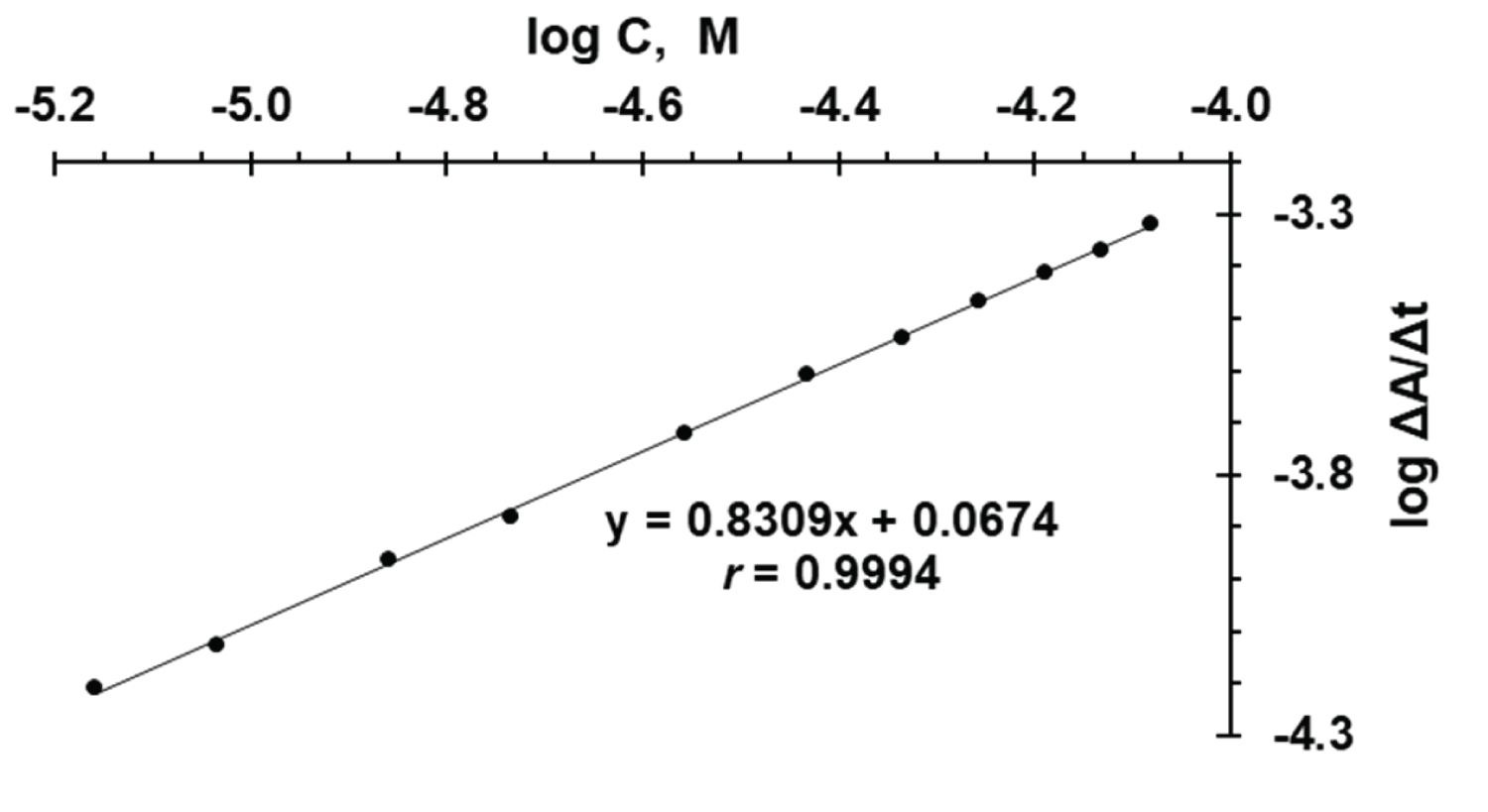
Figure 4: Calibration plot of logarithm....
Calibration plot of logarithm rate (log ν) of the reaction against logarithm molar concentration of FVS for initial rate method.
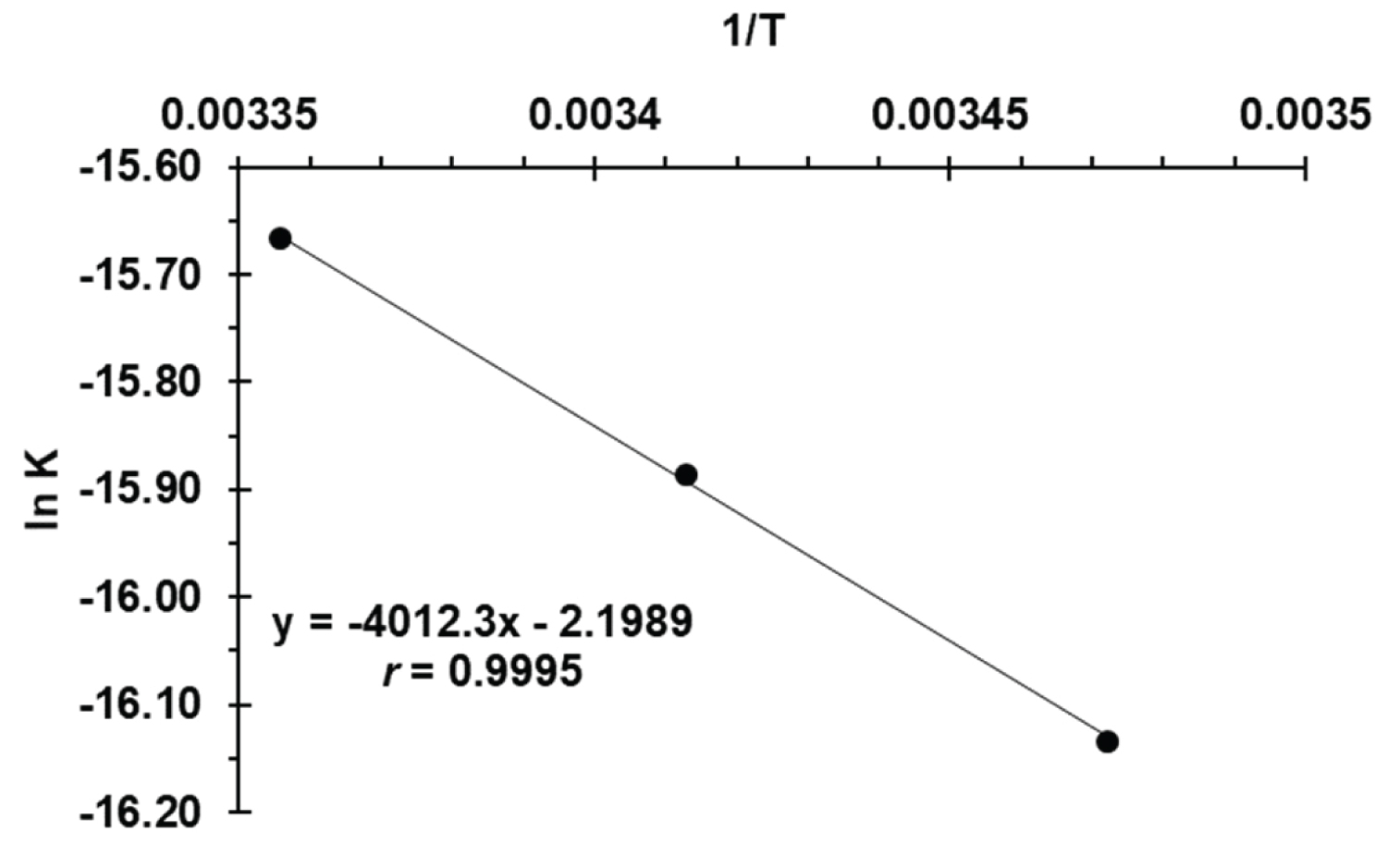
Figure 5: Arrhenius plot of ln k versus....
Arrhenius plot of ln k versus 1/T at 288, 293 and 298 K, CFVS = 7.38 × 10-5 M and CMBTH = 4.0 × 10-4 M for activation energy (Ea).
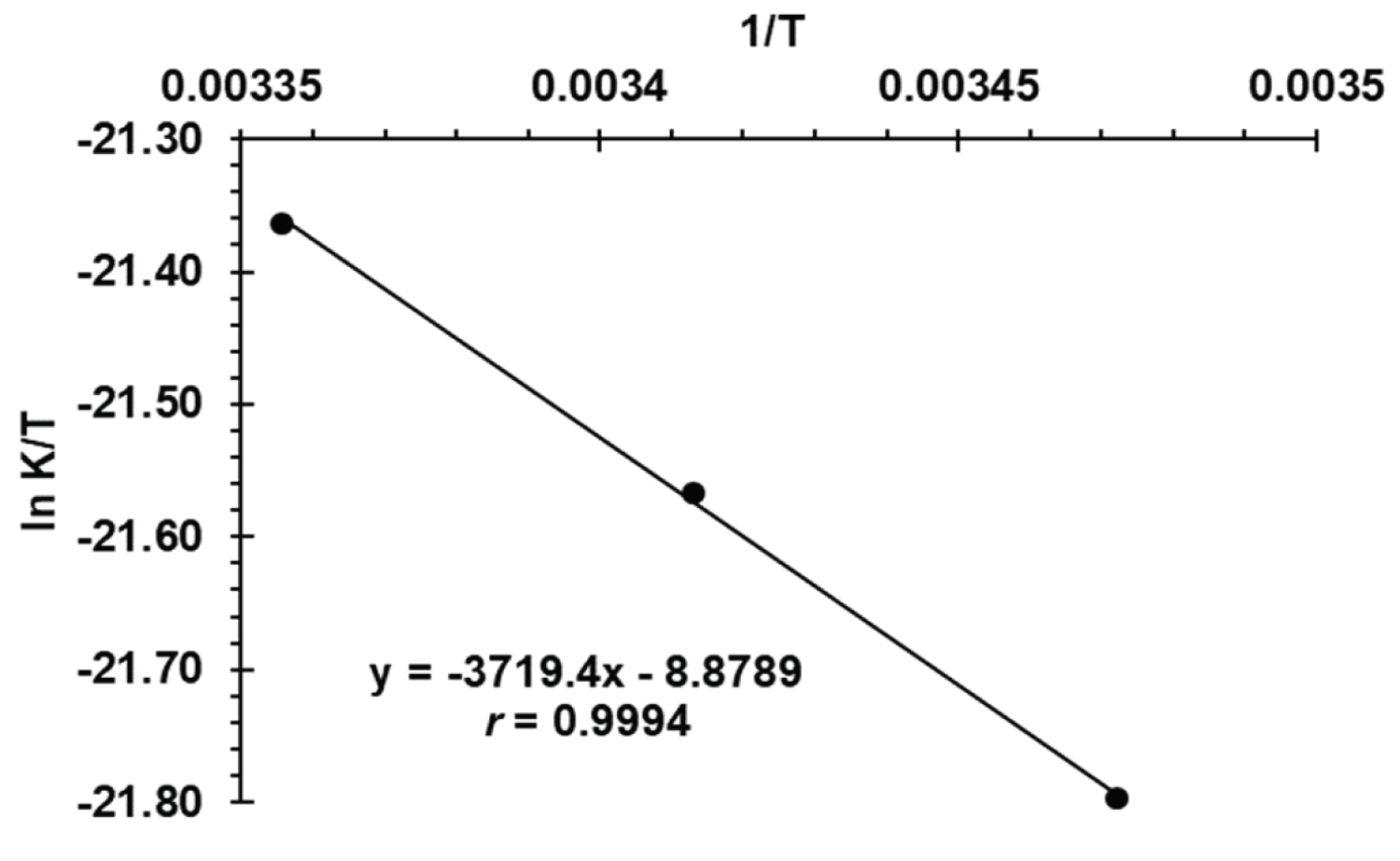
Figure 6: Eyring plot of ln k/T versus....
Eyring plot of ln k/T versus 1/T at 288, 293 and 298 K, CFVS = 7.38 × 10-5 M and CMBTH = 4.0 × 10-4 M.
Tables
Table 1: Analytical parameters for the kinetic determination of FVS using initial rate method at 25 °C.
Table 2: Regression equations for FVS at fixed time and 25 °C.
Table 3: Analytical characteristics of the fixed time (15 min) method.
Table 4: Accuracy and precision for the determination of FVS.
Table 5: Results of robustness study of FVS.
Table 6: Kinetic determination of FVS in capsule dosage forms by the proposed and reference methods.
References
- Walsh P (2005) Physicians’ Desk Reference (PDR). In: Montvale NJ, Medical Economics Company Co. 2326-2331.
- Martindale (2005) The complete drug reference. Sweetman SC, The Pharmaceutical Press, London, UK, 918.
- United States pharmacopeial convention (2021) USP 43-NF 38 the United States pharmacopeia and national formulary. USA.
- British Pharmacopoeia (2013) Her majesty stationery office. London, UK.
- Neves MMPS, Nouws HPA, Delerue-Matos C (2008) Direct electroanalytical determination of fluvastatin in a pharmaceutical dosage form: Batch and flow analysis. Analytical Letters 41: 2794-2804.
- Dogan B, Tuncel S, Uslu B, Özkan SA (2007) Selective electrochemical behavior of highly conductive boron-doped diamond electrodes for fluvastatin sodium oxidation. Diamond & Related Materials 16: 1695-1704.
- Özkan SA, Uslu B (2002) Electrochemical study of fluvastatin sodium analytical application to pharmaceutical dosage forms, human serum, and simulated gastric juice. Anal Bioanal Chem 372: 582-586.
- Yan JL (2006) Determination of fluvastatin sodium by differential pulse voltammetry. Pakistan Journal of Biological Sciences 9: 2156-2158.
- Moussa BA, Abadi AH, Abou-Youssef HE, Mahrouse MA (2010) Spectroscopic and chromatographic determination of fluvastatin sodium in presence of its acid degradate. Int J PharmTech Res 2: 875-898.
- Assassi AL, Roy CE, Perovitch P, Auzerie J, Hamon T, et al. (2015) Green analytical method development for statin analysis. J Chromatogr A 1380: 104-111.
- Ashour S, Khateeb M (2023) New HPLC method for determination of fluvastatin anti-hyperlipidemic drug in bulk and pharmaceutical dosage using simvastatin as internal standard. Drug Analytical Research 7: 21-26.
- Akabari AH, Suhagia BN, Saralai MG, Sutariya VA (2017) Development and validation of stability indicating RP-HPLC method for estimation of fluvastatin sodium in bulk and capsule dosage form. Eurasian Journal of Analytical Chemistry 12: 87-105.
- Gomes FP, García PL, Alves JMP, Singh AK, Kedor-Hackmann ERM, et al. (2009) Development and validation of stability-indicating HPLC methods for quantitative determination of pravastatin, fluvastatin, atorvastatin and rosuvastatin in pharmaceuticals. Analytical Letters 42: 1784-1804.
- Saminathan J, Sankar ASK, Anandakumar K, Srinivasan S, Vetrichelvan T (2009) Validated RP-HPLC method for fluvastatin sodium in bulk and its dosage form. Journal of Pharmaceutical Sciences and Research 1: 90-96.
- Aslan SS, Sagirli O, Ersoy L (2009) Development and validation of a GC-FID assay for determination of fluvastatin in pharmaceutical preparations. Quimica Nova 32: 2347-2350.
- Pesez M, Batros J (1974) Colorimetric and fluorimetric analysis of organic compounds and drugs. Marcel Dekker, New York, 672.
- Long GL, Winefordner JD (1983) Limit of detection, a closer look at the IUPAC definition. Analytical Chemistry 55: 712A-721A.
- Rose J (1964) Advanced physico-chemical experiments. London, Pittman, 67.
- Maree S, Du Preez JL, Du Plessis LH, Du Plessis J, Gerber M (2020) A novel HPLC method developed and validated for the detection and quantification of atorvastatin, fluvastatin, pitavastatin and pravastatin during transdermal delivery studies. Pharmazie 75: 164-166.
- Akabari AH, Shah U, Solanki S, Patel MB, Suhagia BN (2014) Development and validation of sensitive HPTLC method for quantitative analysis of fluvastatin sodium in bulk and pharmaceutical dosage form. International Journal of Pharmaceutical Research 6: 73-78.
- Ashour S, Bahbouh M, Khateeb M (2011) A novel use of oxidative coupling reactions for determination of some statins (cholesterol-lowering drugs) in pharmaceutical formulations. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 78: 913-917.
- Ergin Kizilçay G, Ertürk Toker S (2020) Spectrophotometric determinations of most commonly used statins in pharmaceutical preparations with 2,3-dichloro-5,6-dicyanobenzoquinone. Istanbul J Pharm 50: 95-102.
- Bandla J, Jabeen N, Gorja A (2021) Development and validation of first order derivative UV-spectrophotometric method for the determination of fluvastatin sodium in formulation. International Journal of Pharmaceutical Science and Research 12: 6721-6724.
- Kamal MF, Morshedy S, Saad DA, Moneeb MS (2021) Green atomic absorption spectroscopic methods for the determination of rabeprazole sodium and fluvastatin sodium in pure and pharmaceutical dosage forms. International Research Journal of Pure & Applied Chemistry 22: 8-14.
- Dogrukol-AKD, Kircali K, Tunçel M, Aboul-Enein HY (2001) Validated analysis of fluvastatin in a pharmaceutical formulation and serum by capillary electrophoresis. Biomed Chromatogr 15: 389-392.
- Erk N (2002) Rapid spectrophotometric method for quantitative determination of simvastatin and fluvastatin in human serum and pharmaceutical formulations. Pharmazie 57: 817-819.
- Stolarczyk M, Maslanka A, Apola A, Rybak W, Krzek J (2015) Derivative spectrophotometric method for simultaneous determination of zofenopril and fluvastatin in mixtures and pharmaceutical dosage forms. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 148: 66-71.
- Stolarczyk M, Apola A, Maslanka A, Kwiecien A, Opoka W (2017) Spectrophotometric method for simultaneous determination of valsartan and substances from the group of statins in binary mixtures. Acta Pharm 67: 463-478.
- El-Brashy A, Eid M, Talaat W (2006) Kinetic Spectrophotometric method for the determination of ketoprofen in pharmaceuticals and biological fluids. Int J Biomed Sci 2: 406-413.
- Kopanica M, Satra V, Echschlager K, Rorsak I, Koduys Z, et al. (1983) Kinetic methods in chemical analysis. Elsevier, Amsterdam, The Netherlands, 25-27.
- Pérez-Bendito D, Silva M (1988) Kinetic methods in analytical chemistry. John Wiley and sons, New York, USA, 44-45.
- Atkins P, de Paula J (2006) Physical chemistry for the life sciences. Oxford University Press, NY, USA, 256-259.
- Ashour S, Bayram R (2015) Development and validation of sensitive kinetic spectrophotometric method for the determination of moxifloxacin antibiotic in pure and commercial tablets. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 140: 216-222.
- Ashour S, Bayram R (2019) Selective and validated kinetic spectrophotometric method for the determination of irbesartan in pure and pharmaceutical formulations. Annales Pharmaceutiques Françaises 77: 101-111.
- Ashour S, Bayram R (2020) New sensitive derivatization methodology of levofloxacin antibiotic from its dosage formulations: kinetic spectrophotometric methods for determination. Analytical Chemistry Letters 10: 562-576.
- Ashour S, Bahbouh M, Khateeb M (2010) Kinetic spectrophotometric determination of fluvastatin in pharmaceutical preparations. Int J Biomed Sci 6: 19-26.
- ICH, Q2 (R1) (2005) Validation of analytical procedures: Text and methodology. International conference on harmonization, Geneva.
- Miller JN, Miller JC (2005) Statistics and chemometrics for analytical chemistry. (5 th edn), Chapman & Hall/CRC, London, UK.
Author Details
Safwan Ashour1* and Mouhammed Khateeb2
1Department of Materials Science, Engineering Faculty, Gaziantep University, Gaziantep, Türkiye
2Department of Basic Sciences, Engineering Faculty, Al-Baath University, Homs, Syria
Corresponding author
Safwan Ashour, Department of Materials Science, Engineering Faculty, Gaziantep University, Gaziantep, Türkiye.
Accepted: November 07, 2023 | Published Online: November 09, 2023
Citation: Ashour S, Khateeb M (2023) A Novel Use of Oxidative Coupling Reactions Part II. Kinetic Spectrophotometric Method for Determination of Fluvastatin in Pharmaceutical Formulations. Int J Exp Spectroscopic Tech 8:034
Copyright: © 2023 Ashour S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
A selective kinetic spectrophotometric method has been described for the determination of fluvastatin sodium (FVS) in pure form and capsules. The developed method is based on the oxidative coupling reaction of FVS with 3-methyl-2-benzothiazolinone hydrazone hydrochloride monohydrate (MBTH) in the presence of Ce(IV) in an acidic medium to form colored product followed spectrophotometrically by measuring the increase in absorbance with time at 615 nm. The initial rates, rate constant, fixed absorbance and fixed time methods have been tested for constructing the calibration graphs, and are linear. The most suitable analytical methods were the initial rate and fixed time (at 15 min) methods for determination of FVS in the concentration range of 3.0-36.0 μg/mL with LOD is 0.44 and 0.23 μg/mL, respectively. The activation energy ( E a ), activation enthalpy (D H ‡ ), activation entropy (D S ‡ ) and Gibbs free energy (D G ‡ ) are estimated for the reaction and found to be 33.36 KJ/mol, 30.92 KJ/mol, -271.37 J/K mol and 111.79 KJ/mol, respectively. The kinetic proposed methods have been further applied to the determination of FVS in capsule formulations and the results tallied well with the label claim. The results were statistically compared with those of a reference method by applying the Student's t-test and F-test. No interference was observed from the concomitant substances normally added to preparations.
Keywords
Fluvastatin, Kinetic spectrophotometry, 3-methyl-2-benzothiazolinone hydrazone hydrochloride monohydrate (MBTH), Capsules
Introduction
Fluvastatin sodium (FVS), (3R,5S,6E)-rel-7-[3-(4-fluorophenyl)-1-(1-methylethyl)-1H-indol-2-yl]-3,5-dihyroxy-6-heptenoicacid, monosodium salt, is a competitive inhibitor of HMG-CoA reductase, which is responsible for the conversion of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) to mevalonate, a precursor of sterols including cholesterol. It is used to reduce triglycerides, LDL-cholesterol, apoliporotein B and to increase HDL-cholesterol, in the treatment of hyperlipidaemias including hypercholesterolaemias and combined hyperlipidaemia [1,2]. Fluvastatin sodium and its formulations are official in USP and BP. The USP [3] described HPLC method and BP [4] recommended a non-aqueous titration method for the assay of the drug.
Fluvastatin has been determined, either alone or in combination with other drugs, in pharmaceutical formulations with several analytical methods. These analytical methods include voltammetry [5-8], spectrofluorimetry [9], reversed-phase high performance liquid chromatography (RP-HPLC) with ultraviolet detection [9-15], high performance thin-layer chromatography (HPTLC) [9,16], spectrophotometry [17-19], atomic absorption spectroscopy [20], capillary electrophoresis [21], gas chromatography-FID [22] and derivative spectrophotometry [9,23-25].
Kinetic spectrophotometry using the different reagents is the best choice of the analytical chemist because of its high sensitivity, selectivity and low limit of detection.3-Methyl-2-benzothiazolinonehydrazone hydrochloride (MBTH) is one of the widely used chromogenic reagents for spectrophotometric analysis of phenols. It undergoes an interesting reaction with phenolic, amino, ketonic and aldehydic compounds in the presence of oxidizing agent yielding highly colored reaction products [26]. MBTH had been used for kinetic spectrophotometric determination of many drugs [27-30].
The literature is still poor in analytical assay methods based on kinetics for the determination of fluvastatin in dosage forms. One kinetic spectrophotometric method was found which is based on the formation of yellow product between fluvastatin and 4-chloro-7-nitrobenzofurazan (NBD-Cl) in acetone medium at 55 ± 2 °C [31]. Therefore, there is a need for another kinetic approach more sensitive for determining the content of fluvastatin in pharmaceutical formulations based on the reaction of MBTH with amino group of fluvastatin molecule to form green compound.
Experimental
Instrumentations
A Jasco V-530 UV-VIS spectrophotometer (Japan) with 1 cm quartz cells was used for all absorbance measurements under the following operating conditions: Scan speed medium (400 nm/min), scan range 500-800 nm and slit width 2 nm. Spectra were automatically obtained by Jasco system software. A digimed pH meter (model T-901) with combined glass pH electrode and a Thornton sonicator (model T-14) were used.
Materials and reagent solutions
Working reference standard of fluvastatin sodium (FVS) sample (C 24 H 25 FNO 4 Na, 433.46 g/mole) was supplied by ALPHARM Chemical Co (China). Its purity was 99.6% according to BP [4]. Fluvastatin capsules (Kimi, Syria) labeled to contain 20 mg fluvastatin sodium/capsule and Almastatin capsules (Alma Company, Syria) labeled to contain 40 mg fluvastatin sodium/capsule. 3-methyl-2-benzothiazolinone hydrazone hydrochloride monohydrate (MBTH) 97% was from Aldrich Chemical Co., St. Louis (USA) and the other chemicals and reagents used were of analytical grade and purchased from Merck (Germany). Double distilled water was used in all experiments. 1 × 10 -2 M MBTH solution was prepared in doubly distilled water and Ce(SO 4 ) 2 solution (1% w/v) was prepared with sulfuric acid (0.368 M) medium. Freshly prepared solutions were always used.
Standard solutions of FVS
A stock standard solution 0.5 mg/mL of FVS was prepared by dissolving the appropriate weight of fluvastatin sodium in distilled water. Working standard solutions were then prepared by suitable dilution of the stock standard solution with doubly distilled water. The stability of the solutions was studied by measuring UV absorption spectra and found to be stable for three days when stored in the dark at 2-8 °C.
Recommended kinetic procedures for determination of FVS
Aliquots of standard FVS (0.15-1.80 mL and 0.20-1.60 mL of 0.5 mg/mL for initial rate and fixed time methods, respectively) solutions were transferred into a series of 25 mL calibrated volumetric flasks. 1.0 mL of 1 × 10 -2 M MBTH solution was added and kept aside for 7 min. After that, 1.0 mL of 1% w/v Ce(SO 4 ) 2 solution was added. The volume was made up to the mark with distilled water and shaken well.
In the initial rate procedure, the absorbance-time plot was recorded at 615 nm against reagent blank at 25 ± 2 °C. The initial rate of the reaction at different concentrations was obtained from the slope of the tangent to the absorbance time curve. A calibration curve was constructed by plotting the logarithm of the initial rate of the reaction versus the logarithm of the molar concentration of FVS. In the fixed time procedure, the absorbance measured at a fixed time 15 min was plotted against the final concentration of FVS. The content of FVS was computed from regression equations of the calibration graphs.
Proposed kinetic methods validation
The method was validated in accordance with the International Conference on Harmonization (ICH) guidelines [32]. The following validation characteristics were addressed:
Linearity: The calibration graph was constructed applying the general assay procedure using either the initial rate or fixed time method for the determination of FVS.
Sensitivity: The sensitivity of the method was determined on the limit of detection (LOD) and limit of quantification (LOQ). LOD and LOQ of the FVS assay were determined experimentally after getting the regression equation. LOD and LOQ were calculated by the following equations: and (where b is the slope of the calibration curve and is the standard deviation of the intercept) [33].
Precision: The intraday precision was determined by measuring FVS samples of 4.0-36.0 μg/mL measured five times on the same day. The percentage relative standard deviation (RSD%) of the assay values was calculated.
Accuracy: The percentage relative error (Er%) and recovery were calculated in view to justify the accuracy of the proposed method.
Selectivity: The selectivity of the method was ascertained by analyzing standard FVS in the presence of common excipients such as lactose, magnesium stearate and cellulose. For this purpose, a powder blend using typical tablet excipients was prepared along with the drug and then analyzed.
Robustness: The robustness of an analytical procedure is a measure of its capacity to remain unaffected by small, but deliberate variations in method parameters and provides an indication of its reliability during normal usage. These condition changes included the concentration of the oxidant and reagent in the reaction and the time of measurement.
Procedure for the determination of FVS in capsules
The entire content of twenty capsules containing FVS were weighed and pulverized. An amount of the powder equivalent to 25 mg of FVS was dissolved in a 25 mL of methanol and mixed for about 5 min and then filtered through Whatman filter paper number 40. The methanol was evaporated to the dryness. The remaining portion of solution was diluted in a 50 mL volumetric flask to the volume with double distilled water to achieve a concentration of 0.5 mg/mL. The recommended kinetic procedures were followed within the concentration range achieved above.
Results and Discussion
In our previous work [17], MBTH has been used for the spectrophotometric determination of some statins (atorvastatin, fluvastatin and pravastatin) in pure and in dosage forms. The reaction of MBTH with FVS in the presence of Ce(SO 4 ) 2 in an acidic medium at 25 °C was resulted in the formation of an intense green-colored oxidative coupling product that exhibits an absorption maximum at 615 nm (Figure 1). The pure drug and the reagent blank (MBTH) showed a negligible absorbance at the corresponding maximum at 615 nm. Several parameters such as reagent concentrations, time, temperature and sequence of additions were optimized to achieve high sensitivity, low blank reading and high stability for the determination of FVS.1.0 mL of 10 -2 M MBTH and 1.0 mL of 1%w/v Ce(SO 4 ) 2 in 0.368 M H 2 SO 4 medium were the optirnum volumes for the maximal absorbance readings for such reaction to form the oxidative coupling product with high intense colour.Addition of FVS, MBTH and Ce(SO 4 ) 2 in that order gave maximum absorbance.
The effect of temperature on the initial rate of reaction of FVS with MBTH in acid medium was studied in the range 288-303 K. With an increase in temperature, FVS reacts faster with MBTH. At 303 K and higher temperatures up to 313 K, the reaction product remained stable. For the sake of good results, 298 K was selected as the optimum temperature for the determination process.The influence of the reaction time was investigated in the range of 0-30 min. The experimental results show that the final color was achieved with 10 min and the color product was stable for at least two hours up to 40 °C and was measured at 615 nm.
Stoichiometry
The limiting logarithmic method [34] was used to evaluate the stoichiometry of FVS-MBTH complex. Two sets of solutions were performed. In the first one, MBTH concentration was varied keeping a constant concentration of FVS, while in the second, MBTH concentration was kept constant and FVS concentration was varied. The order of reaction of FVS with respect to MBTH or vice versa was determined by plotting log absorbance versus log [MBTH] or log [FVS] to estimate the slope of the respective line (Figure 2a and Figure 2b). By dividing the slopes of MBTH curve (0.7809 and 1.7847) over the slope of the FVS curve (0.7810) the ratio of the reaction was calculated and found to be 1:1 and 1:2 FVS to MBTH. We also found from Job’s method of continuous variation and mole-ratiothat a molar ratio is 1:1 and 1:2 FVS to MBTH with the formation constant ( K f ) value of FVS-MBTH complex is 7.42 and 8.04 at ratio 1:1 and 1:2, respectively [17].
Quantitation kinetic methods
The absorbance-time plot for the reaction of FVS with MBTH in the presence of Ce(SO 4 ) 2 in H 2 SO 4 medium is investigated at 615 nm and 25 ± 2 °C. The reaction was completed within 15 min and after that became stable, so the 15 min development time was optimally chosen in the general procedure (Figure 3). The absorption of the chromogen remains stable for at least two hours. It is well known that the most common kinetic analytical methods are the initial rate, rate constant, variable time (fixed concentration or fixed absorbance) and fixed time methods [35,36]. These methods have been tested in order to select the most appropriate analytical methods with a high sensitivity value and the higher values of the correlation coefficient ( r ).
Initial rate method: The initial reaction rate obeys the following equation:
ν = ΔA/Δt = k C n
Where ν is the reaction rate, A is the absorbance, t is the measuring time, k is the pseudo order rate constant, C is the concentration of the drug mol/L and n (slope) is the order of the reaction. The above equation is transformed into the logarithmic form as follows:
log ν = log ΔA/Δt = n log C + log k
The regression of logarithm of the initial rate of reaction (log ν ) versus logarithm of drug concentrations (log C ) gave a linear regression equation (1), as shown in Figure 4 and Table 1,
log ν = 0.8309 log C + 0.0674 (1)
Over the concentration range of 6.92 × 10 -6 - 8.31 × 10 -5 M FVS (3.0-36.0 μg/mL) with correlation coefficient ( r = 0.9994) and slope value of n = 0.8309 describing a first order rate with respect to FVS concentration, Thus, log k = 0.0674 and then k = 1.77 S -1 . The limit of detection (LOD) and limit of quantification (LOQ) for initial rate method were determined and were found to be 0.44 and 1.33 μg/mL, respectively.
The apparent activation parameters such as the activation energy ( E a ) activation enthalpy (D H ‡ ), activation entropy (D S ‡ ) and Gibbs free energy (D G ‡ ) can be estimated by study the reaction rate at different temperatures and fixing the reaction conditions [37]. The activation energy of the reaction under investigation can be determined from the logarithmic form of Arrhenius equation (2):
(2)
Where k is the rate, A is a constant known as Arrhenius frequency factor, E a is the activation energy, T is the absolute temperature (273 °C) and R is the gas constant (8.3143 J/K mol). The absorbance of the reaction product are measured at different temperatures; 15, 20 and 25 °C and keeping the concentrations of C FVS = 7.38 × 10 -5 M and C MBTH = 4.0 × 10 -4 M constant in acid medium. The absorbance-time plot at these temperatures was constructed to determine the rates, and then the Arrhenius plot of lnk obs. versus 1/T was found to be linear with a correlation coefficient of -0.9995 (Figure 5). The value of the line slope was: (- E a /R) = -4.0123 × 10 3 k and the activation energy of the investigated product was: E a = 33.36 K J/mol. The positive sign of the activation energy indicated that the product formation increased with increasing the temperature.
The value of D H ‡ and D S ‡ were evaluated for the reaction from the Eyring equation (3):
Where k b is Boltzman’s constant (1.381 × 10 -23 J/K) and h is Blank’s constant (6.626 × 10 -34 JS). The plot of lnk obs. /T versus 1/T was linear with a correlation coefficient of -0.9994 (Figure 6). The value of D H ‡ and D S ‡ were evaluated from the slope (-D H /R) and the intercept [ln(k b /h) + D S /R] of Eyring plot and found to be 30.92 KJ/mol and -271.37 J/K mol, respectively. The value of Gibbs free energy (D G ‡ ) of activation of the reaction product was determined using the equation (4), and its value was 111.79 KJ/mol. Since D H ‡ > 0 and D S ‡ < 0 this leads to Δ G ‡ > 0 which indicates non-spontaneous, endothermic condensation reaction and tends toward regularity.
Δ G ‡ = D H ‡ - T D S ‡ (4)
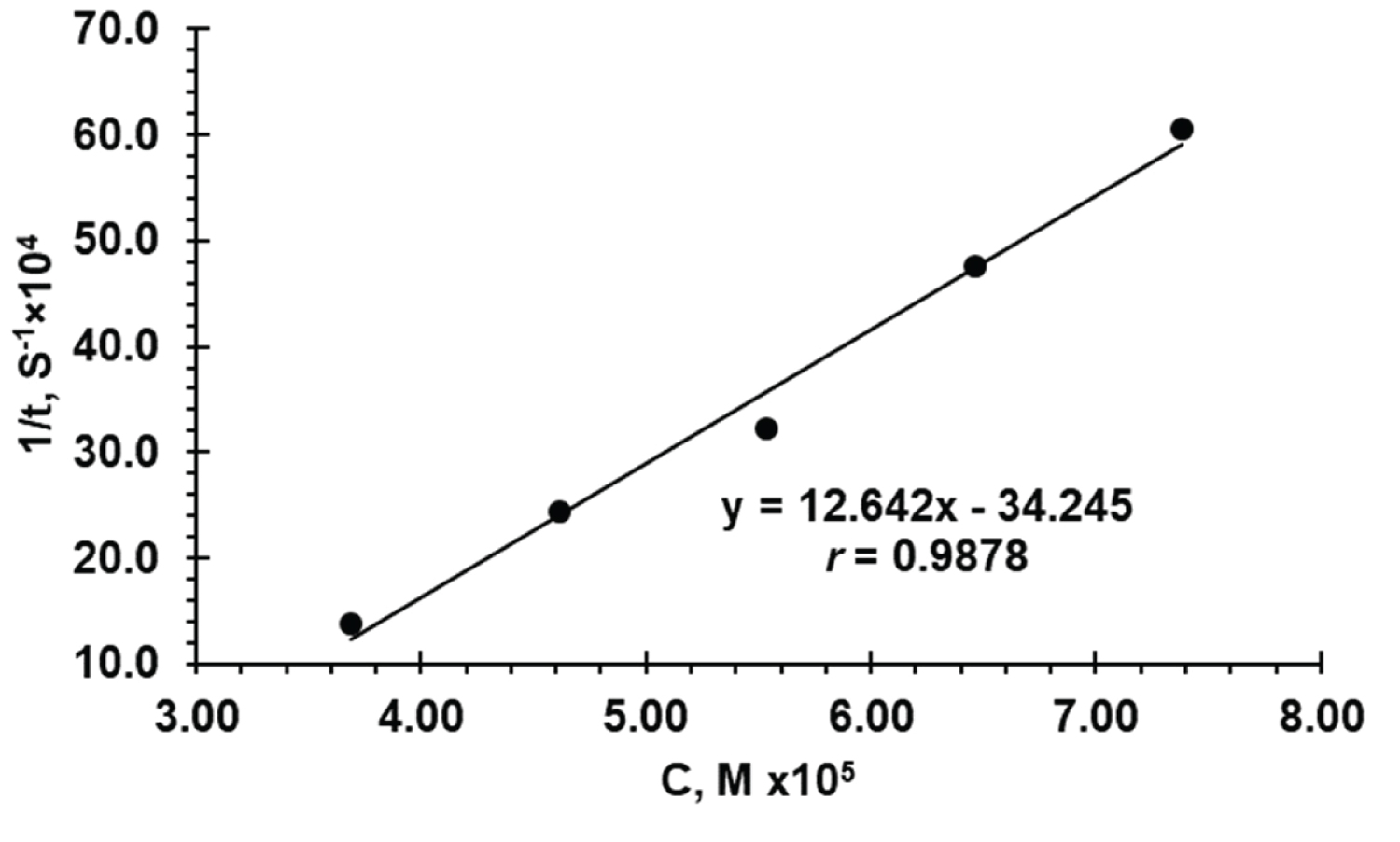
Fixed absorbance method : Reaction rate data were recorded for different FVS concentrations in the range 4.0-36.0 μg/mL (0.92 × 10 -5 - 8.30 × 10 -5 M). A preselected value of the absorbance 0.38 was fixed and the time was measured in the seconds. The reciprocal of time (1/ t ) versus the initial concentration of FVS was plotted (Figure 7) and the equation (5) of calibration graph was obtained:
1/ t = 12.624 C - 0.0034254 (5)
The range of FVS concentrations giving the most satisfactory results was limited 16.0-32.0 μg/mL (3.69 × 10 -5 - 7.38 × 10 -5 M) with correlation coefficient of r = 0.9878.
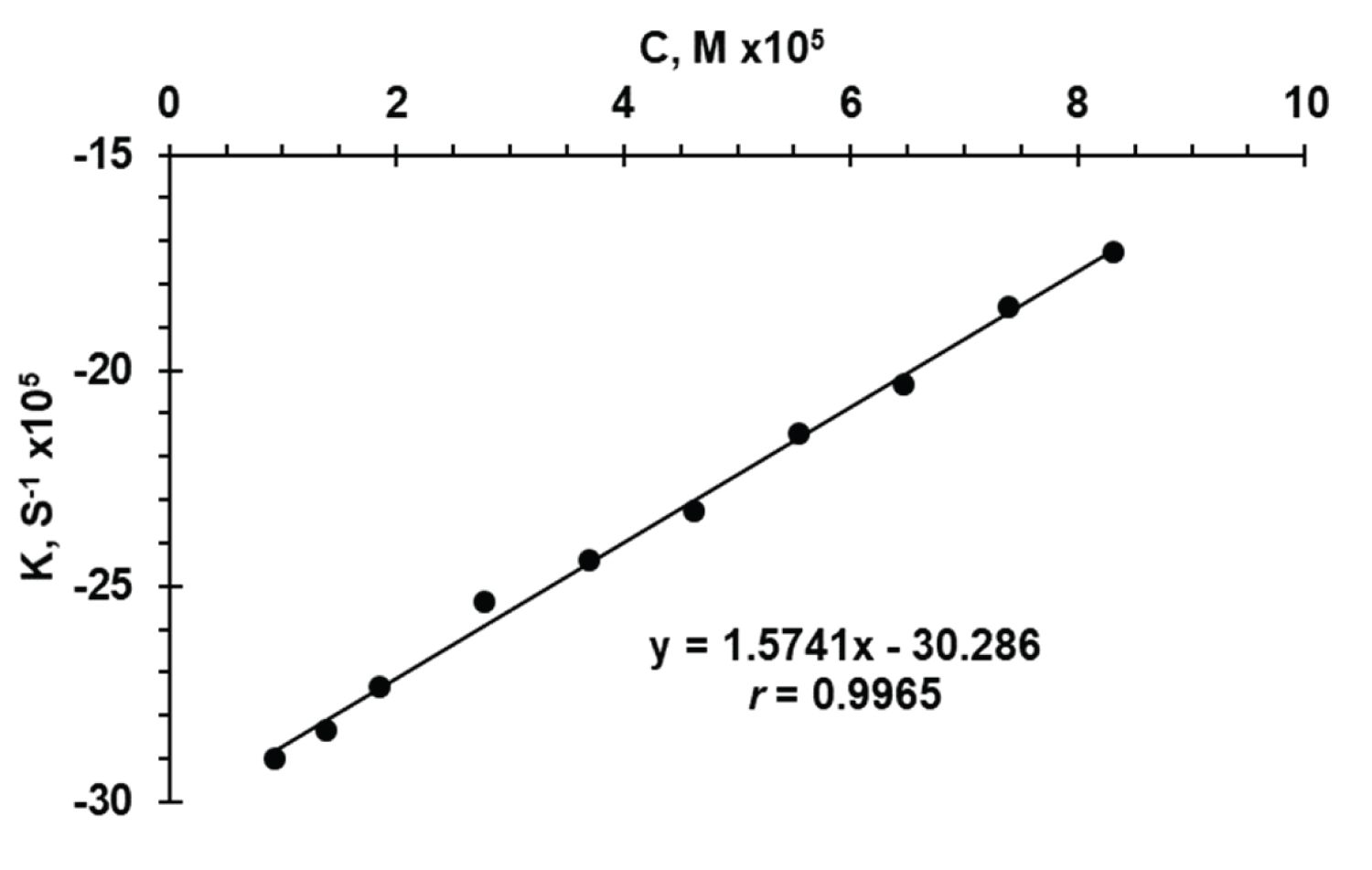
Rate constant method: Graphs of log A versus time for FVS concentrations in the range of 3.0-36.0 μg/mL were plotted and all appeared to be rectilinear. Pseudo order rate constant (k) corresponding to different FVS concentrations were calculated from the slopes multiplied by -2.303. Regression of C versus k (Figure 8) gave the equation (6), and was linear in the range of 4.0-36.0 μg/mL (0.92 × 10 -5 - 8.30 × 10 -5 M) with r = 0.9965. The value of r indicates poor linearity which is probably due to inconsistency of k .
k = 1.5741 C - 0.00030286 (6)
Fixed time method: In this method, a preselected fixed time is chosen and the absorbance of orange colored solutions containing varying amounts of FVS was measured at this time. The absorbance against the initial concentrations of FVS was plotted at fixed time 5, 10, 15, 20, 25 and 30 min to construct the calibration graphs. The least squares method was used to calculate the correlation coefficient, intercept and slope values for the regression equations of these calibration curves [38]. Table 2 shows the calibration data, linear ranges and the optimum time is 15 min to obtain the most acceptable values of linearity and correlation coefficient. Therefore, the fixed time of 15 min was selected for the determination of FVS in pharmaceutical preparations.
Validation of the proposed kinetic methods
Linearity, LOD and LOQ: After setting the optimal conditions for the reaction, it was found that the most acceptable correlation coefficient values are for the initial rate and fixed time (15 min) methods, which were chosen for the validation study. The calibration graph for the initial rate or fixed time method was constructed by applying the general assay procedure for determination of FVS. Regression equation of initial rate method depending on initial rates of the FVS reaction was equation (1) with r = 0.9994. The linear range was 3.0-36.0 μg/mL with LOD and LOQ of 0.44 μg/mL and 1.33 μg/mL, respectively. In the fixed time method, the most appropriate fixed time that corresponds to the most acceptable values of the correlation coefficient was 15 min and chosen for all measurements. The regression equation for FVS, linear range, correlation coefficients, Ringbom optimal concentration range, LOD, LOQ and Sandell sensitivity were calculated and reported in Table 3.
Accuracy and precision: The accuracy and precision of the proposed methods have been evaluated by measuring six replicate analyses at five different concentrations of FVS on the same day. The precision of the established methods was assessed by calculating the percentage relative standard deviation (RSD%). The accuracy was evaluated by calculating the percentage relative error (Er%) and recovery. The low values of RSD% and Er% indicate the high precision and the good accuracy of the proposed methods. The percentage recovery was found to be within range. The results tabulated in Table 4 demonstrated excellent recoveries with low values of standard deviations. The data also showed that no interference from the common excipients used in capsules. This method has a striking feature that is its relative freedom from interference by the usual capsule additives and excipients in amounts far exceeding what may naturally occur in pharmaceutical preparations.
Selectivity: The selectivity of the method was ascertained by analyzing standard FVS in the presence of common excipients such as lactose, magnesium stearate and cellulose. For this purpose, a powder blend using typical tablet excipients was prepared along with the drug and then analyzed. The proposed method was found to be selective for the determination of FVS in the presence of various capsules excipients. The recoveries were not affected by the excipients and the excipients blend did not show any absorption in the range of analysis.
Robustness: The conditions of the proposed methods for the estimation of FVS in pure and capsules were studied and are robust. The operational robust parameters that are investigated were as shows in Table 5. The robustness of the methods under these conditions for sample solutions containing 24.0 µg/mL of FVS was studied by measuring five replicates. The degree of reproducibility of the results obtained as a result of small deliberate variations in the method parameters has proven that the proposed methods are robust.
Analysis of fluvastatin in capsule dosage forms
The kinetic proposed methods were successfully applied to the determination of FVS in commercial capsules. The results of the initial rate and fixed time methods for the assay FVS in capsules were compared with that of the reference method [4] and are listed in Table 6. Mean values were obtained with a Student’s t- and F-tests at 95% confidence limit for five degrees of freedom [38]. The calculated t- and F-values do not exceed the theoretical ones indicating no significant differences between the kinetic proposed methods and the reference method. The average percent recoveries obtained were quantitative indicating good accuracy of method. The results showed that the method is suitable for routine analysis of drugs in formulations.
Conclusion
The developed kinetic spectrophotometric method is the first kinetic method for the determination of fluvastatin with MBTH reagent in acid medium. It is quite simple, robust, do not requires any pretreatment of the drug, higher accurate and sensitive as compared to similar reported methods [31], selective and hence can be used for the routine analysis of fluvastatin in bulk and pharmaceutical formulations with a limit of detection of 0.23 mg/mL. The sample recoveries from the formulation were in good agreement with their respective label claims, which suggested non-interference of formulations excipients in the estimation. Moreover, the lower reagents consumption leads to an environmentally friendly spectrophotometric procedure, which makes it especially suitable for routine quality control analysis work and research laboratories.
Competing Interests
All of the authors declare that they have no competing interests.